
 Español
Español  Português
Português  English
English Dr. Fernando L. Soldano, Dr. Antonio Molina Rojas y Dr. Gustavo Lavenia
Síndrome caracterizado por proteinuria (signo capital), edemas, disproteinemias (hipoalbuminemia, hipergammaglobulinemia), y dislipemia. Es la consecuencia clínica del aumento de la permeabilidad de la pared capilar glomerular, por lo tanto, es una manifestación de enfermedad glomerular renal. Se considera que una proteinuria puede producir síndrome nefrótico si es superior a 3,5 g/ 24 h/1,73 m2 en adultos o 40 mg/h/m2 en niños (proteinuria en rango nefrótico). Sin embargo, las manifestaciones clínicas del síndrome nefrótico pueden no aparecer con cifras superiores a este valor o ser vistas con cifras menores de 3,5 g/24 h. La importancia del síndrome nefrótico está en la magnitud de la proteinuria, y en la lesión glomerular subyacente, por lo tanto, la proteinuria es un factor de mal pronóstico evolutivo de la lesión renal y de la enfermedad cardiovascular.
Fisiopatología de la proteinuria del síndrome nefrótico
La fisiopatología central del síndrome nefrótico radica en el aumento de la permeabilidad glomerular y la consiguiente pérdida de proteínas por la orina. El resto de las alteraciones que se describen en el síndrome nefrótico son consecuencia directa de esta proteinuria masiva.
La pared capilar glomerular normal, compuesta por las células endoteliales, la membrana basal glomerular y las células epiteliales viscerales, constituye una barrera por tamaño y por carga eléctrica al paso de proteínas de tamaño superior a 70 kd. En el síndrome nefrótico, la proteinuria podría producirse, al menos teóricamente, por un trastorno electroquímico (pérdida de la electronegatividad de la barrera de filtración), o bien por una alteración estructural de la membrana de filtración que condicione un aumento del tamaño de los poros de la misma. En tamaño de los poros en la pared de las células epiteliales viscerales, tiene un papel fundamental, ya que evitarían el paso de macromoléculas de tamaño superior a 150 kd. Diversas moléculas de adhesión, como la nefrina, así como integrinas y proteínas del citoesqueleto, parecen ser constituyentes fundamentales de estos poros. Además, en algunos modelos teóricos se sugiere la existencia de otros poros de mayor radio, cuyo número estaría aumentado en el daño glomerular, favoreciendo la proteinuria masiva. Estos poros disminuirían con fármacos antiproteinúricos como los inhibidores de la enzima conversora de angiotensina (IECA). La alteración estructural de las células epiteliales glomerulares es característica de todos los procesos que cursan con síndrome nefrótico. La limitada capacidad proliferativa de estas células tras una noxa favorecería la proteinuria y la insudación de proteínas plasmáticas que mediante microscopia óptica se traduciría en depósitos proteicos, alteraciones en la membrana basal glomerular y anomalías del mesangio glomerular.
La barrera en función de la carga se debe a las cargas negativas de los glucosaminoglucanos polianiónicos ricos en heparán sulfato de la membrana basal glomerular, que restringirían el paso de pequeñas proteínas polianiónicas plasmáticas de tamaño entre 70-150 kd, principalmente, la albúmina. Por esto, las proteínas cargadas positivamente, a igualdad de tamaños, presentan un mayor aclaramiento renal respecto a las de carga negativa. La nefropatía de cambios mínimos constituye el paradigma de las enfermedades causadas por un trastorno glomerular electroquímico. En estos casos no se observan alteraciones morfológicas con microscopia óptica, y la proteinuria es muy selectiva (se pierden, sobre todo, albúmina y otras proteínas negativas, mientras que aquellas de mayor peso molecular, como la IgG, quedan retenidas). La causa de este aumento de la permeabilidad glomerular se desconoce, pero se piensa en un factor circulante liberado por linfocitos y monocitos.
Pequeñas modificaciones en las propiedades de permeabilidad de la pared glomerular producen importantes pérdidas de proteínas de peso molecular intermedio (entre 40 y 150 kd). Entre las que se incluyen: albúmina, IgG, transferrina, ceruloplasmina y glucoproteína. También se pierden pequeñas cantidades de proteínas de tamaño algo superior como las formas pequeñas de HDL (200 kd). Las proteínas de muy elevado peso molecular como IgM, macroglobulinas, fibrinógeno, factor XIII, fibronectina y lipoproteínas de mayor tamaño no se pierden, incluso con grandes alteraciones en la permeabilidad y selectividad glomerular. Desde un punto de vista práctico, en el síndrome nefrótico no se pierden proteínas de tamaño superior a los 200 kd.
Independientemente de la extensión del daño glomerular, otros factores pueden condicionar una variación muy amplia en la proteinuria, como son el filtrado glomerular, el flujo plasmático renal, la actividad del sistema renina-angiotensina, la producción y concentración plasmática de albúmina, la ingesta proteica diaria y la administración de fármacos antihipertensivos.
Clínica del Síndrome Nefrótico
Hipoalbuminemia
La albúmina es la proteína plasmática más abundante. La albúmina filtrada es catabolizada en parte por el túbulo renal, cuya tasa catabólica aumenta, pudiendo representar hasta un 20% de la albúmina filtrada en el síndrome nefrótico.
Para compensar las pérdidas, el hígado aumenta la tasa de síntesis de albúmina hasta en un 300% por mecanismos de transcripción. Este incremento se correlaciona con la albuminuria, pero no con la presión oncótica del plasma o con la concentración sérica de albúmina, y se abole si la ingesta proteica está disminuida, lo que explica por qué las dietas hipoproteicas disminuyen la proteinuria, pero no aumentan la concentración de albúmina en el plasma.
La hipoalbuminemia (albúmina inferior a 3 g/dl) aparece cuando la proteinuria y el catabolismo renal de la albúmina superan la capacidad de síntesis hepática. El grado de hipoalbuminemia se correlaciona con la magnitud de la proteinuria, aunque no de forma constante, ya que otros factores como la edad, el estado nutricional y el tipo de lesión renal también influyen, lo que justifica que haya pacientes con proteinurias muy elevadas sin hipoalbuminemia. Este hallazgo es característico de algunas lesiones glomerulares que cursan con hiperfiltración, como la nefropatía de la obesidad, la nefropatía de reflujo o la secundaria a reducción de masa renal.
Edemas
La aparición de edemas es el signo clínico más visible y suele ser el motivo de consulta, es un edema blando, con fóvea y que se acumula en zonas declives (pies, sacro) y en regiones con presión tisular pequeña, como en la región periorbitaria. Si la hipoalbuminemia es grave puede aparecer ascitis y derrame pleural. El edema pulmonar no se produce a menos que exista insuficiencia renal o cardíaca.
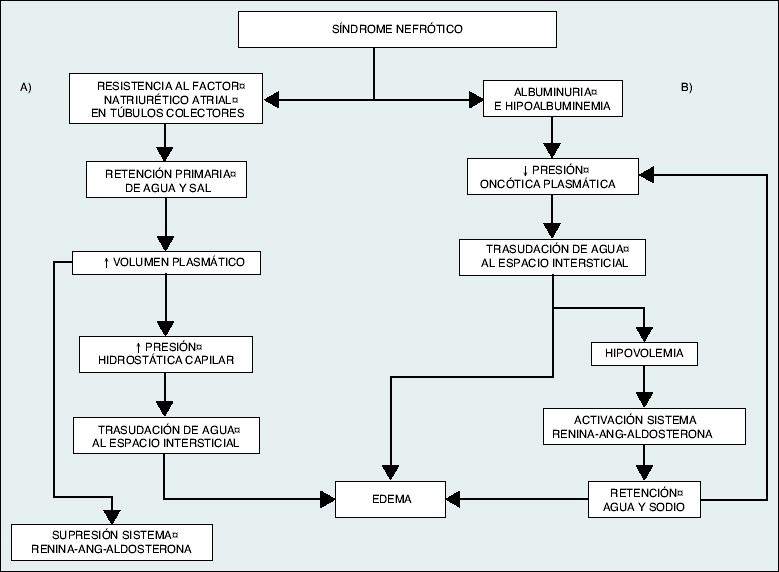
La teoría clásica es la de la hipovolemia, donde la retención renal de agua y sodio es consecuencia de la disminución de la presión oncótica plasmática resultante de la hipoalbuminemia. Se favorecería así la extravasación de líquido del compartimiento intravascular al intersticial, ocasionando edemas. La respuesta homeostática a la hipovolemia activaría el sistema renina-angiotensina-aldosterona, aumentaría el tono simpático y la liberación de hormona antidiurética. Gradualmente, el volumen plasmático se normalizaría a expensas de un aumento del espacio extracelular y de un incremento importante del edema.
La teoría alternativa que explicaría el origen de los edemas, con la teoría de expansión de volumen o hipervolemia. Según ésta, se produciría un daño intrínseco de la nefrona que conduciría a un aumento primario en la reabsorción tubular de sodio, independientemente de la situación hemodinámica, induciéndose una expansión de volumen que favorecería la aparición de edemas. La teoría de la hipervolemia es hoy día la más aceptada. En caso de hipoalbuminemias muy graves, sobre todo en niños, se daría la situación de hipovolemia efectiva y tendrían más trascendencia los mecanismos propuestos por la teoría clásica o de hipovolemia.
|
Patogenia de la formación del edema en el síndrome nefrótico. A) Teoría de la hipervolemia o expansión de volumen, según la cual el edema sería resultado de la retención de agua y sodio por el propio riñón enfermo, que produciría una expansión del volumen plasmático y un aumento en la presión hidrostática capilar que, junto a la hipoalbuminemia, favorecería la trasudación de líquido al espacio intersticial. B) Teoría clásica o de la hipovolemia, según la cual el descenso en la presión oncótica secundario a la hipoalbuminemia favorecería una situación de hipovolemia y la retención de agua y sodio a través de la activación del sistema renina-angiotensina-aldosterona. Esta situación sólo tiene trascendencia clínica en circunstancias de hipoalbuminemia grave (albúmina < 2 g/dl). |
Dislipemia
Es el resultado de un incremento en la síntesis hepática de lípidos y apolipoproteínas y, sobre todo, de un descenso en el aclaramiento de quilomicrones, lipoproteínas de muy baja densidad (VLDL), de baja densidad (LDL) y de densidad intermedia (IDL).
La hipercolesterolemia es la anomalía lipídica más constante y su gravedad se correlaciona de forma inversa con la magnitud de la hipoalbuminemia y con el descenso de la presión oncótica plasmática. La hipertrigliceridemia, menos frecuente, sólo aparece cuando la albúmina sérica disminuye por debajo de 1-2 g/dl.
De forma muy característica existe lipiduria, observándose cilindros grasos en el sedimento de orina. Es recomendable adoptar las medidas terapéuticas necesarias para normalizar la hiperlipemia de todo paciente con síndrome nefrótico, como drogas hipolipemiantes para
Reducir la morbimortalidad cardiovascular o de la progresión de la enfermedad renal con la corrección de las anomalías lipídicas.
Trombosis
La trombosis de la vena renal y, en general, los fenómenos tromboembólicos constituyen una de las complicaciones más graves del síndrome nefrótico. La hipoalbuminemia (albúmina sérica inferior a 2,5 g/dl), la proteinuria superior a 10 g/24 h, valores de antitrombina III inferiores al 75% de lo normal y la hipovolemia se han asociado con un riesgo elevado de complicaciones tromboembólicas.
Infecciones
Hay varios factores que condicionan una elevada susceptibilidad a las infecciones en los pacientes nefróticos, incluyendo déficit de IgG (por disminución de su síntesis y aumento de las pérdidas por filtración y catabolismo renales), opsonización inadecuada por disminución en el factor B del complemento, así como trastornos en la inmunidad celular favorecidos por el déficit de vitamina D, la malnutrición, y las carencias de transferrina y zinc, ambos esenciales para la adecuada función linfocitaria.
Hipertensión arterial
Hallazgo frecuente en el síndrome nefrótico. Los factores predisponentes a la hipertensión no se conocen con claridad, pudiendo estar relacionados con la retención de sodio y agua, o con la pérdida urinaria de sustancias vasodilatadoras (hipotensoras).
Alteraciones endócrinas
Existe un déficit relativo de EPO (Eritropoyetina), sin embargo, la repercusión sobre la eritropoyesis es nula, excepto en casos aislados. Pueden hallarse además alteraciones en el metabolismo de la vitamina D, disminución de las concentraciones de tiroxina (T4) y de triyodotironina total (T3) pero los pacientes están clínicamente eutiroieos.
Otras complicaciones
En los síndromes nefróticos de larga duración pueden aparecer otras complicaciones. Así, la proteinuria persistente y el aumento en el catabolismo renal de las proteínas filtradas desencadenan un balance negativo de nitrógeno y una malnutrición proteica. También se han descrito alteraciones tubulares proximales, como glucosuria, hiperfosfaturia y síndrome de Fanconi.
 Enfermedades glomerulares
Enfermedades glomerulares
Inicialmente, explicaremos cuales son en general, la clasificación de las enfermedades glomerulares por tiempo de evolución y luego por etiología. Referiremos en este capítulo, en especial a las que cursan con Síndrome Nefrótico y a continuación las que cursan predominantemente con Síndrome Nefrítico. Un primer enfoque clínico permite clasificar las glomerulonefritis en función de su evolución en el tiempo. Las glomerulonefritis agudas evolucionan en días, con un comienzo, y muchas veces un fin, bien delimitado en el tiempo. Las glomerulonefritis rápidamente progresivas se caracterizan por un deterioro rápido de la función renal a lo largo de semanas o meses, sin tendencia espontánea a la mejoría, con un sustrato histológico común, que es la proliferación extracapilar en forma de semilunas epiteliales. Las glomerulonefritis crónicas se caracterizan por su curso insidioso y evolución variable a lo largo de los años. Otro enfoque es el etiológico, ya que en muchas de las glomerulonefritis puede identificarse un factor causal. Se distinguen así dos grandes grupos: glomerulonefritis primarias y secundarias. Sin embargo, esta clasificación etiológica tiene muchas limitaciones, ya que no nos permite distinguir entre las distintas glomerulonefritis primarias, cuyo origen es desconocido y, además, una misma causa puede producir varios patrones de enfermedad glomerular de curso clínico y pronóstico distinto. En el siguiente esquema veremos la celularidad e histología glomerular normal, a fin de comprender las diferentes variantes de glomerulopatías que pueden hallarse.
|
Clasificación histológica de las glomerulonefritis primarias
|
|
Clasificación etiológica, histológica y clínica de las glomerulonefritis (GN)
GN asociadas a enfermedades sistémicas
|
Causas del síndrome nefrótico
Cualquier enfermedad glomerular, primaria o secundaria, puede producir síndrome nefrótico en algún momento de su evolución. Las causas más frecuentes se enumeran en tabla 1. Las enfermedades que producen síndrome nefrótico varían con la edad. Así, la mayoría de los síndromes nefróticos en niños se deben a nefropatía de cambios mínimos. En los adultos, la causa más frecuente es una forma secundaria, la nefropatía diabética. La prevalencia de glomerulonefritis primarias, en adultos con síndrome nefrótico, es variable, dependiendo de la región geográfica y de la población estudiada. Las más frecuentes son la nefropatía membranosa, la glomerulonefritis esclerosante y focal (en aumento y la primera causa en la raza negra) y la nefropatía por cambios mínimos.
| Etiología del síndrome nefrótico | ||
| Glomerulonefritis primarias | Niños (%) | Adultos (%) |
| Nefropatía de cambios mínimos | 52.2 | 14.8 |
| Glomerulonefritis esclerosante y focal | 33.3 | 15.1 |
| Glomerulonefritis membranosa | 5.8 | 22.2 |
| Glomerulonefritis mesangiocapilar | 4.3 | 7 |
| Nefropatía por IgA | - | 4.9 |
| Otras lesiones glomerulares primarias | - | 10.3 |
|
Enfermedades glomerulares secundarias
|
|
Evaluación clínica y de laboratorio del síndrome nefrótico
|
Nefropatías glomerulares
Nefrosis Lipoidea o cambios mínimos:
Es una enfermedad típica de la infancia (80 %), aunque puede presentarse a cualquier edad. Clínicamente, se caracteriza por alteraciones de síndrome nefrótico puro, con proteinuria selectiva. Aunque usualmente es idiopático, puede, excepcionalmente, asociarse a otras enfermedades. Escasa alteración funcional, a excepción de su presentación en edades avanzadas, donde pude aparecer insuficiencia renal oligúrica, determinada por la hipovolemia. Histológicamente se caracteriza por la ausencia de lesiones histológicas relevantes por microscopia óptica y presencia de fusión de los podocitos por microscopia electrónica. En la inmunofluorescencia, aunque generalmente es negativa, se puede ver algún depósito mesangial de IgM o complemento, acompañado o no de mínima proliferación mesangial. Es controversia actual si la nefrosis lipoidea y la glomerulonefritis segmentaria y focal son dos entidades distintas o dos facetas de la misma enfermedad, en la actualidad parece prevalecer la idea de que son dos entidades diferentes con distinto comportamiento clínico, pronóstico e incluso etiopatogenia
Glomeruloesclerosis segmentaria y focal:
Puede presentarse en adultos y en niños, puede ser primaria y secundaria. Se caracteriza por la presencia de lesiones esclerosantes en algunos glomérulos (focal) y solo en algunas asas capilares (segmentaria) y depósitos inmunes de IgG y C3. Clínicamente presentan síndrome nefrótico con proteinuria no-selectiva, microhematuria, leucocituria, Hipertensión arterial y grados variables de afectación de la función renal.
Glomerulopatía Membranosa:
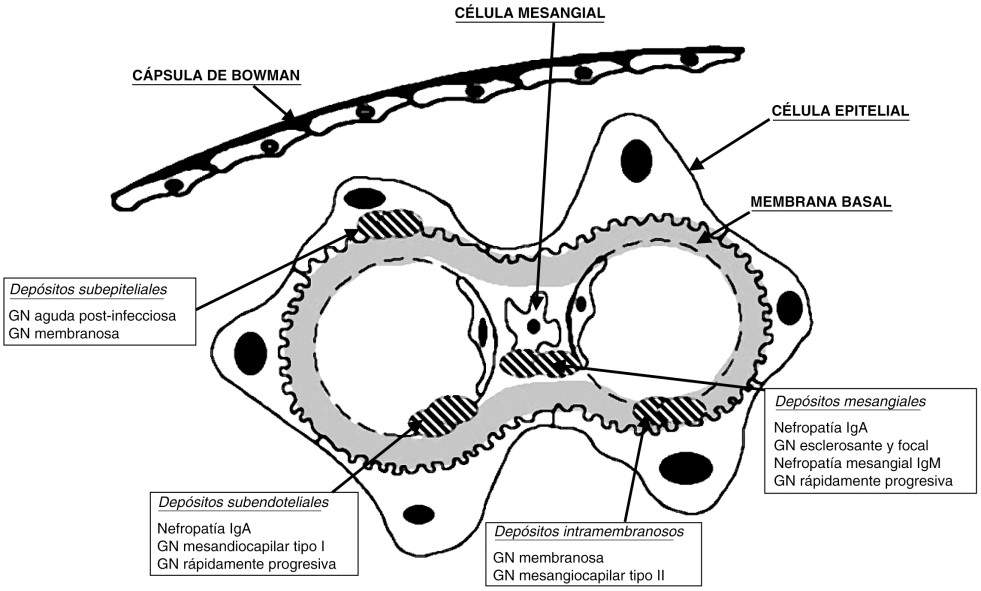
Clínicamente presenta proteinuria intensa y constituye la causa más frecuente de síndrome nefrótico en el adulto. Puede ser primaria y secundaria, en las formas secundarias, pueden asociarse a virus Hepatitis B, C y neoplasias. Desde el punto de vista morfológico, se caracteriza por la existencia de depósitos subepiteliales a lo largo de la membrana basal en ausencia de proliferación celular, lo que da la característica histológica de engrosamiento difuso de la membrana basal. Dicho depósito es gradual y da la clasificación morfológica en 4 estadios. Entre un 20 a 30% de esta glomerulopatía es autolimitante. A medida que progresa la lesión, los glomérulos se esclerosan y se produce fibrosis intersticial y atrofia tubular, lo que conduce a IRC en años.
Glomerulopatía Membranoproliferativa:
También conocida como mesangiocapilar, se trata de una enfermedad glomerular crónica, puede ser primaria y secundaria, de curso clínico variable. En las formas secundarias, se encuentran como factores, enfermedades infecciosas, hereditarias o multisistémicas (LES, Hepatitis, neoplasias, crioglobulinemia, etc.). La característica predominante es la proliferación mesangial, junto con engrosamiento de la pared capilar glomerular e hipocomplementemia (característica la disminución de C3). Existen 3 tipos según la clasificación histológica: Tipo I, forma clásica, con depósitos subendoteliales; tipo II, con depósitos densos intramembranosos; Tipo III, depósitos subendoteliales y subepiteliales. La presentación clínica no difiere en los subtipos, predomina síndrome nefrótico (50%) asociado a hematuria y/o anormalidades urinarias asintomáticas. Un alto porcentaje evolucionan a IRC y presenta recidiva en el Trasplante Renal.
Glomerulopatía Proliferativa Mesangial Difusa:
Poco frecuente, histológicamente de caracteriza por lesiones mesangiales no específicas (incremento de la celularidad mesangial en forma difusa y lesiones de esclerosis glomerular) y clínicamente cursa con síndrome nefrótico con microhematuria. En la inmunofluorescencia, puede hallarse depósitos de IgM, IgA y C3.
Glomerulonefritis Rápidamente Evolutiva:
Se define clínicamente por rápido deterioro de la función renal en semanas o meses, con escasa tendencia a la recuperación espontánea. La característica histológica es la proliferación extracapilar en forma de semilunas superior al 50%, su incidencia es variable y puede producirse en cualquier edad. Existen formas primarias (idiopáticas, con 3 tipos) y secundarias a enfermedades autoinmunes e infecciosas.