
 English
English  Português
Português  Español
Español Julio Libman, Astrid L. Libman
The congenital adrenal hyperplasia syndrome, whose clinical expression varies according to its various forms, is due to the total or partial deficiency of some of the enzymes involved in the synthesis of adrenal steroids, with a decrease in the production of steroids distal to the blockage and increase of some previous to it, by accumulation and deviation of the synthesis towards alternative metabolic pathways.
Pathophysiology
Congenital adrenal hyperplasia constitutes a group of autosomal recessive alterations in adrenal steroidogenesis.
The synthesis of adrenal steroids occurs from cholesterol by the action of a number of enzymes (Fig. 66-1). Its production and secretion, especially that of glucocorticoids and sexually acting steroids, and to a much lesser extent that of aldosterone, is regulated by CNS centers, which control the production of the hypothalamic ACTH-releasing hormone, a 41 amino acid peptide that Through the hypothalamic-pituitary portal system, it reaches the adenohypophysis, where it determines the release of ACTH, a 39 amino acid peptide whose main physiological effect is to stimulate the synthesis of cortisol. ACTH secretion, which occurs episodically, and cortisol secretion are subject to what is known as the circadian rhythm, with maximum levels in the morning hours and minimum in the last hours of the day.
The blockage of cortisol synthesis due to the total or partial lack of any of the enzymes involved in its synthesis, causes a decrease in its plasma levels, an increase in ACTH production and bilateral adrenal hyperplasia. When the enzyme deficiency is marked, the production of cortisol is not normalized, but that of the precursors with androgenic action or of Na + retention increases significantly, which determines a series of variable clinical manifestations according to the location of the enzyme blockade.
Symptoms and signs
The manifestations of the clinical picture vary according to the enzyme deficiency present.
21-hydroxylase deficiency . It is the most common type of congenital adrenal hyperplasia. There are two varieties: a) the simple form and b) the salt-losing form.
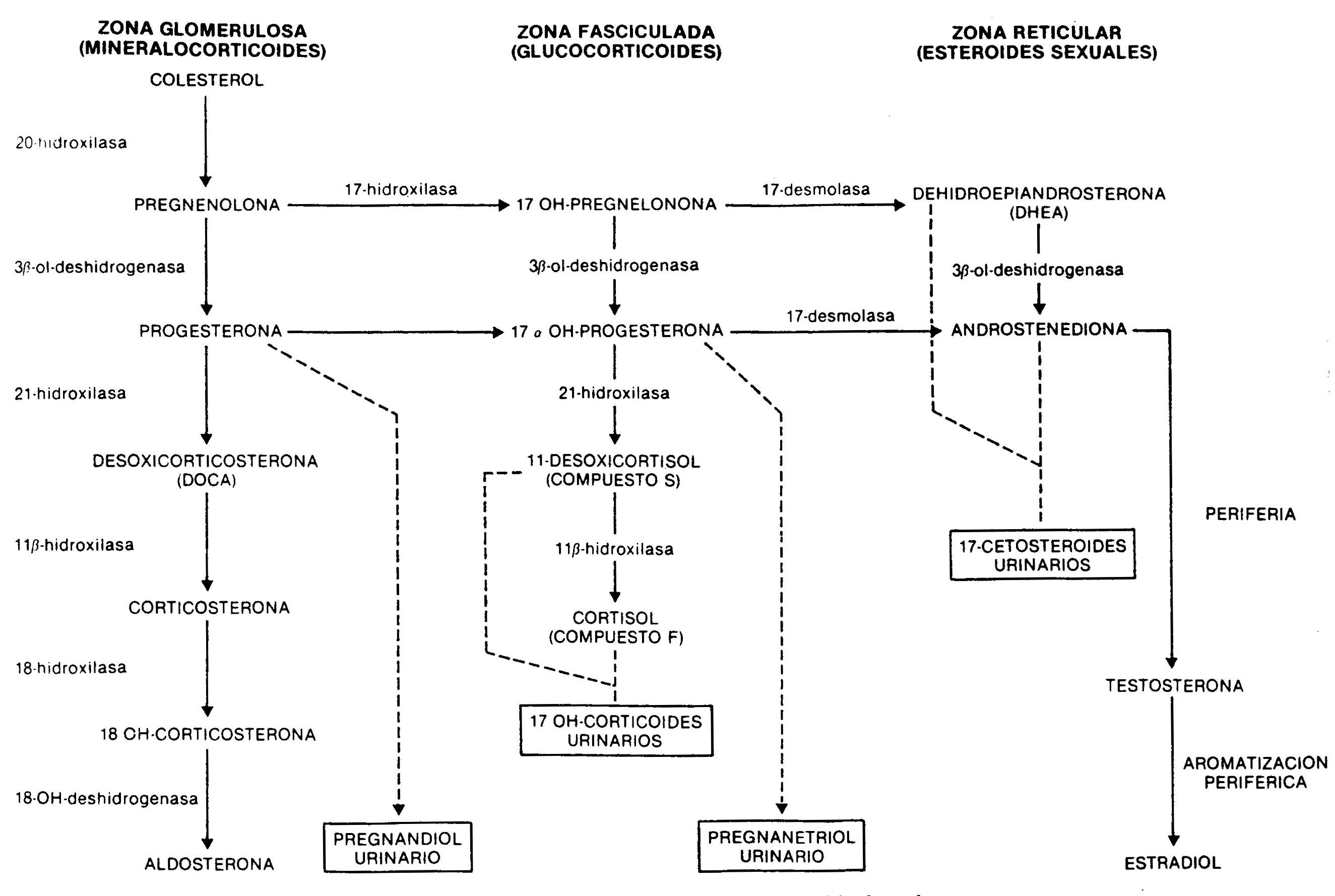
Fig. 66.1 - Synthesis of adrenal steroids from cholesterol
a) In the simple variety there is a partial deficiency well compensated for by an increase in ACTH, so that the production of cortisol approaches normality. The increase in ACTH produces a marked elevation of androgens and cortisol precursors. Androstenedione, highly increased in the blood, is a weak androgen that is metabolized peripherally to testosterone; this causes masculinization of the external genitalia during fetal life in the genotypically female patient, with clitoral enlargement and varying degrees of fusion of the labia majora, which can mimic a male with cryptorchidism and hypospadias. In males there are no abnormalities in the external genitalia at birth. Subsequently, a progressive virilization occurs in both sexes, with the appearance of axillary and pubic hair, facial, changes in the voice and increased muscle mass. Even when in early childhood they are taller than that corresponding to age and sex, early fusion of the epiphyses due to androgenic action leads to short final stature. At puberty, and in the absence of adequate treatment, the physiological increase in gonadotropins does not occur due to an inhibition by circulating sex steroids, and the result of this is primary amenorrhea in women and small testes in men. In the simple, non-losing form of salt, there are no difficulties in the hydrosaline balance and the blood pressure is normal. 17-ketosteroids, 17OH-progesterone and, to a lesser extent, pregnandiol are increased, decreasing their values in urine after administration for 7 days of 1,
b) In the salt-losing form there is a complete deficiency of 21-hydroxylase. Even when there is a compensatory increase in ACTH, cortisol levels are low, as are aldosterone, so that signs of acute adrenal insufficiency appear within the first week of life, with anorexia, vomiting, loss of weight, diarrhea, dehydration and hyperkalemia. If left untreated, vascular collapse develops and death ensues.
Prenatal diagnosis of 21-hydroxylase deficiency can be made in the first trimester of pregnancy by molecular genetic analysis of chorionic villus cells and quantitation of 17-hydroxyprogesterone in a small amount of amniotic fluid.
11-beta-hydroxylase deficiency. It causes a deficiency in cortisol synthesis with hypersecretion of ACTH. The height of the block determines the excessive production of cortisol precursors, such as 11-deoxycortisol and its tetrahydro-derivative metabolite, which are quantified in the urine as 17OH-corticosteroids. Consequently, these are elevated, as are 17OH-progesterone and its metabolite, pregnanetriol. The deficiency also limits the conversion of deoxycorticosterone to corticosterone, with the consequent accumulation of this potent mineralocorticoid, responsible for the arterial hypertension seen in this form of congenital adrenal hyperplasia. As a consequence of the enzymatic deficit, the short circuit towards the synthesis of androgens also occurs. 11-hydroxylase deficiency is therefore characterized by
20-hydroxylase or cholesterol desmolase deficiency . It is a rare form that affects the conversion of cholesterol to pregnenolone; consequently, there is an alteration in the production of glucocorticoids, mineralocorticoids, and sex steroids. The lack of the former determines a severe picture of adrenal insufficiency, with loss of Na +, dehydration, hypovolemia and hypotension. Androgen deficiency is the cause of affected patients having female external genitalia, whether their karyotype is XX or XY.
3-beta-ol-dehydrogenase deficiency . There is a defect in the conversion of pregnenolone to progesterone, from 17OH-pregnenolone to 17OH-progesterone, and from dehydroepiandrosterone to androstenedione. As a consequence, there is a decrease in mineralocorticoids, glucocorticoids, and testosterone, which causes adrenal insufficiency with ambiguous genitalia in men and moderate clitoral hypertrophy in women, due to the accumulation of dehydroepiandrosterone, a weak androgen. This defect is also present in the gonad.
17-hydroxylase deficiency . This enzyme deficiency results in the total or partial inability to synthesize cortisol and sex steroids. As seen in Figure 66-1, this enzyme is not involved in the production of mineralocorticoids. The production of deoxycorticosterone, controlled by ACTH, is greatly increased, resulting in mild arterial hypertension, Na + retention, expansion of plasma volume, and inhibition of the renin-angiotensin system. The increased production of corticosterone compensates for the lower synthesis of cortisol. Males show ambiguous genitalia due to decreased testosterone production. Enzyme deficiency is also present in the ovaries, with the consequent lack of sexual development at puberty.
Study methodology
From the above, it can be inferred that the existence of congenital adrenal hyperplasia in a newborn with ambiguous external genitalia can be suspected, associated or not with vomiting, dehydration and circulatory collapse, or the development of hypertension. In an older man, it may present with accelerated growth and precocious pseudopuberty, that is, development of secondary sexual characteristics with infantile testicles.
In the presence of some of these manifestations, nuclear chromatin should be evaluated in a cell smear of the buccal mucosa and in circulating leukocytes in order to determine chromosomal sex, and to quantify urinary 17-ketosteroids, 17OH-corticosteroids and pregnanetriol, and 17OH-progesterone and blood ionogram, together with 11-deoxycortisol, in case of concomitant hypertension. Bone age is advanced on hand and wrist radiographic images.