
 English
English  Português
Português  Español
Español Carlos J. Galli Mainini
Adams-Stokes syndrome . see Stokes-Adams syndrome
Adie syndrome . Neurological syndrome of unknown etiology. It is characterized by a pathological reaction of the pupil to accommodation (myotonia), in which the pupil on the affected side contracts and dilates more slowly than on the healthy side. Certain tendon reflexes are absent or diminished, but there are no other motor or sensory disorders or signs of central nervous system disease.
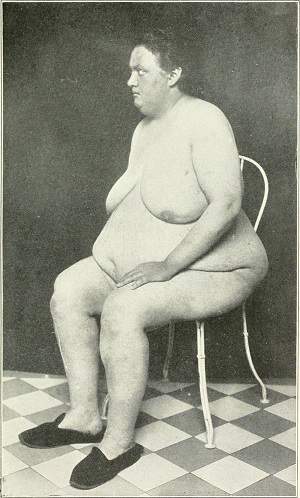
Frohlich's adiposogenital syndrome . Endocrinogenital syndrome due to hypothalamic dysfunction. Its basic features are: a) feminoid obesity with hypoplasia of the gonads: and absence of secondary sexual characteristics; b) diabetes insipidus; and c) mental and growth retardation and vision disorders. It is produced by tumors or other pathological processes that affect the hypothalamic pituitary area and that include the hypothalamic center of appetite, responsible for obesity.
Adrenogenital syndrome . Set of clinical pictures produced by adrenal hyperfunction and whose manifestations are predominantly genital. It includes the following: a) congenital adrenal hyperplasia syndrome. caused by specific deficiency of enzymes, which are involved in corticosteroid metabolism (see Chap. 61); b) the adrenal virilization syndrome in adults, produced by benign or malignant adrenal tumors, and c) the adrenal feminization syndrome, caused by tumors or by enzyme deficiency.
Motor aphasia syndrome . Neurological syndrome due to occlusion of the superior division of the middle cerebral artery. The classical picture is characterized by: a) the individual retains his internal language (he knows what to say) but has lost the memory of the movements necessary to do it, so that he cannot express that language in words: articulated (anarthria) ; b) understands what is being said, and reading and writing are preserved; c) is fully aware of what is happening, which often leads to exasperation, and d) in some cases variable disturbances of understanding are associated. It is due to lesions of the inferior frontal, anterior parietal and anterior insular regions, of embolic origin and sometimes due to brain tumors or abscesses.
Sensory aphasia syndrome . Neurological syndrome due to occlusion of the left middle cerebral artery.It results from the loss of the ability to understand the meaning of words (comprehension aphasia), without alterations in the articulation of words. The individual is deprived of all means of social communication. There is: a) inability of the patient to make himself understood (neither verbally nor in writing), because the correct words of the language are replaced by others of more or less similar sound or meaning (literal and verbal paraphasias), or by neologisms or jargon incomprehensible; b) inability to find the name of the objects it perceives; c) inability to understand what he reads or what is said to him, and d) feeling, on the part of the observer, that the patient does not have a clear awareness of what is happening to him.
 Frohlich's 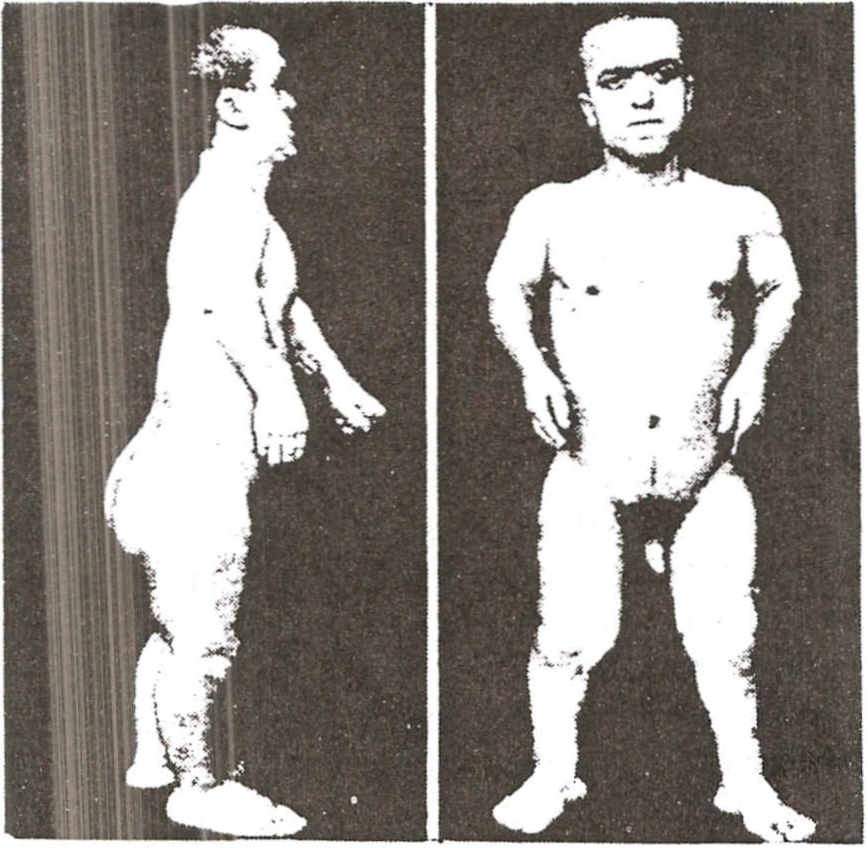 syndrome Achondroplasia syndrome (Suros, Medical Semiology, 1968) |
Visual agnosia syndrome . Neuro-ophthalmological syndrome of central origin. It is produced by injuries that affect the visual pathways and the primary, secondary, and tertiary cortical areas of vision, and the picture is that of an individual who sees objects well, but cannot recognize them unless, according to the case, touch, smell or taste them, or recognize the sounds they make.
Optic hole syndrome . Neurological syndrome of the base of the skull. Unilateral blindness, insidious onset and progressive in nature, accompanied or not by ipsilateral pain radiating to the neck and mandibular region. It is caused, almost always, by a glioma of the optic nerve.
Anterior ragged hole syndrome . see Bonnet syndrome
Posterior ragged hole syndrome . see Vernet Syndrome
Ahumada-Del Castillo syndrome . see Forbes-Albright syndrome
Sphenoid wing minor syndrome . see Kennedy syndrome
Albright-McCune-Sternberg syndrome . Hereditary syndrome characterized by bone, skin and endocrine-gynecological disorders. It is a rare condition seen in girls, and is manifested by bone dysplasia (thickening, curving and easy fractures of the bones), pigmentary abnormalities of the skin (spots with irregular edges, coffee-with-milk color) and precocious puberty with early development secondary sexual characteristics.
Fetal alcohol syndrome . Infant syndrome due to alcohol abuse by the mother during pregnancy. The following traits may be present, isolated or in combination: a) short stature in relation to that which would correspond according to weight; b) facial (hypoplasia of the lower jaw, cleft palate) and ocular (microphthalmia, epicanthus fold) and bilateral hip dislocation; c) cardiac and genital anomalies, and d) cardiac and renal malformations. In addition, the newborn can develop a similar picture; only that it is persistent, that of abstinence from alcohol in adults, and with time it shows a mental retardation of variable degree.
Alexia syndrome without agraphia . Neurological syndrome due to occipital lobe injury. Right hemianopia and inability to read aloud and understand written language, without alterations in dictation or spontaneous reading in silence. It is due to a lesion of the left occipital lobe, usually tumor, associated with a lesion of the corpus callosum.
 Albright-McCune-Sternberg syndrome Albright-McCune-Sternberg syndrome
|
Andersen syndrome . Complicated mucoviscidosis syndrome. It is a triad of mucoviscidosis (cystic fibrosis of the pancreas), bronchiectasis and hypovitaminosis A, in which the first component is the underlying disease, and the other two are frequent complications.
Cerebellopontine angle syndrome . Central neurological syndrome with involvement of cranial nerves V, VII and VIII. It is manifested by perception deafness (CN VIII), followed by labyrinthine areflexia or hyporeflexia (CN VIII), and sometimes associated with corneal areflexia (CN V) and facial paralysis of variable intensity (CN VII). It is caused by meningiomas or neurinomas of the acoustic nerve.
Anxiety syndrome . Functional syndrome of anxiety states. Paleness, sweating, variable tremor, palpitations, rapid and shallow breathing or "short" breathing, unwarranted fear, etc., with no detectable organic cause. It is observed in individuals subjected to sustained situations of psychic tension.
Crush syndrome . Syndrome due crushing ' held in any sector of the body. It defines the picture of edema, oliguria, hypovolemia, shock, and finally, acute renal failure, which is frequently observed in individuals who have suffered prolonged traumatic compression injuries, particularly when the trauma has affected a considerable mass of muscle tissue (p. g., crushing of both lower limbs as a result of a collapse).
Sleep apnea syndrome . see Hypersomnia syndrome with nocturnal apnea
Superior mesenteric artery syndrome . Abdominal syndrome caused by extrinsic obstruction of the duodenal transit. It is caused by the superior mesenteric artery by compressing the third portion of the duodenum against the trunk of the abdominal aorta, as a consequence of which an acute and transient obstruction, but recurrent in time, of the segment in question occurs. The manifestations of the syndrome are highly variable: from minimal disturbances to postprandial nausea and vomiting, central abdominal pain, and occasionally extreme distention of the stomach and duodenum.
Afferent loop syndrome . Syndrome secondary to gastrojejunostomy. It is manifested by recurrent postprandial episodes of nausea and bloating and pain in the upper abdomen. The condition is attributed to intermittent chronic partial obstruction of the proximal intestinal loop (duodenum and jejunum)
Blind loop syndrome . Small bowel dysfunction syndrome. It is characterized, in its severe forms, by steatorrhea, poor absorption of vitamin B 12 and non-iron deficiency anemia. It is attributed to abnormal bacterial proliferation of the small intestine, as a consequence of stenosis of the organ or of operations that leave intestinal loops with little active transit.
Ashernan syndrome . Endometrial destruction amenorrhea syndrome. Secondary amenorrhea and sterility, with intrauterine scars and adhesions, and more or less complete disappearance of the endometrium. It is caused by instrumental procedures (curettage), usually from postpartum hemorrhage or abortion complicated by infection.
Double athetosis syndrome . see Vogt syndrome .
Aubertin syndrome . Syndrome of abrupt interruption of blood flow in the heart chambers. It is a picture, of sudden and rapidly progressive appearance of acute retrosternal pain, dyspnea, tachypnea, anguish, lividity, threadlike pulse, collapse and almost always death, which is observed as a complication of mitral stenosis and occurs when a clot detaches from the left atrium abruptly occludes the atrioventricular foramen.
Auriculotemporal syndrome . see Frey's syndrome .
Avellis syndrome . Neurological syndrome of the brainstem with motor and sensory alterations, and involvement of the X cranial nerve. Vocal cord and soft palate paralysis on the side of the lesion (X cranial nerve), and on the opposite side hemiplegia respecting the face and loss of thermal and pain sensitivity (lesion of the roof of the bulb and the pyramidal bundle). The cause is tumor or brain softening.
Ayerza syndrome . Advanced chronic pulmonary failure syndrome Severe cyanosis, congestive heart failure, and varying degrees of polycythemia.
Babinski-Nageotteo syndrome . Neurological syndrome of unilateral medulla oblongata. Hemiplegia and hemianesthesia on the opposite side of the injury, and hemiasynergia, hemiataxia, and lateropulsion on the same side of the injury; and if the sympathetic fibers of the reticular formation are affected, also miosis, enophthalmia and palpebral ptosis on the side of the lesion. It is generally due to a vascular obstruction above the crossing of the pyramidal bundle.
Babinski-Vaquez syndrome . Neurological and vascular syndrome of advanced syphilis. It is a variable and polymorphic condition, which depends on the location and severity of the main lesions and which is manifested, basically, by alteration of the tendon reflexes (patellar and achillian areflexia) and those of the pupil (pupillary areflexia, miosis, et.), anomalies. cerebrospinal fluid, demented state and overt aortic regurgitation.
Bafuerstedt syndrome . Benign cutaneous lymphadenosis syndrome. Inflammatory hyperplasia of the lymphatic follicles of the skin, preferentially affecting the face and ears, in young adults, in the form of solitary or disseminated nodules and which usually regress spontaneously.
Bandl-Frommel syndrome . Syndrome of impending rupture of the uterus. It is typical of the second stage of labor and is characterized by tetanization of the uterine body and the ring of Bandl, and by the possibility of palpating two hard and tense cords that correspond to the round ligaments through the abdominal wall.
Banti syndrome . Chronic congestive splenomegaly syndrome. It is due to processes that produce chronic hypertension of the portal system, with secondary hypersplenism and is characterized by: a) splenomegaly; b) anemia, leukopenia and thrombocytopenia, with hyperplasia of the bone marrow and c) liver cirrhosis, with ascites and hemorrhages due to ruptured esophageal varices.
Bard and Pic syndrome . Pancreas head carcinoma syndrome. It is manifested by a characteristic triad: progressive chronic jaundice, great enlargement of the gallbladder, and rapidly evolving cachexia. It is due to compression of the common bile duct by the tumor mass.
 Barraquer-Simmons Barraquer-Simmons 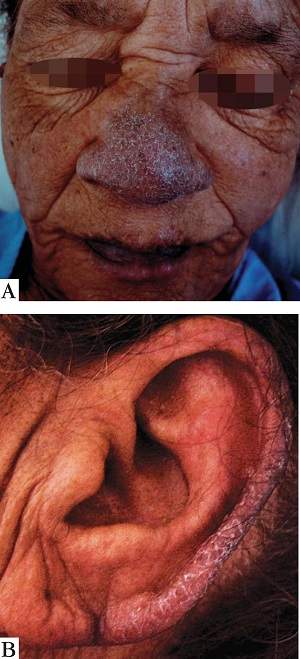 Syndrome Bazex Syndrome |
Barraquer-Simmons syndrome . Progressive lipodystrophy syndrome. Deep wasting of the upper part of the body, particularly the face, and great obesity in the lower half, especially the buttocks and thighs. It is seen in young women and the cause is unknown.
Barré-Lieou syndrome . Syndrome related to organic conditions of the cervical spine. It is characterized by headache, temporary visual disturbances, vasomotor phenomena in the face, vertigo, tinnitus, craniofacial paresthesia, intermittent dysphagia and dysphonia, etc. It is caused by direct irritation of the spinal and sympathetic nerves, in patients with advanced osteoarthritis of the last three, cervical vertebrae, or by vascular (vertebrobasilar) or psychogenic disorders.
Barrett's syndrome . Chronic esophagitis syndrome. Chronic inflammation of the mucosa of the lower third of the esophagus, associated or not with the development of peptic ulcers, which progressively leads to stenosis due to scar retraction. The underlying cause is, in general, the replacement of the squamous or squamous epithelium of the esophagus. by a gastric-type secretory epithelium.
Bartter syndrome . Secondary hyperaldosteronism syndrome with juxtaglomerular cell hyperplasia. It is characterized by hypokalemic alkalosis and a marked increase in plasma renin levels, without arterial hypertension or response to the pressor action of angiotensin. The cause is probably genetic and the condition may be accompanied by mental retardation and short stature.
Bassen-Kornzweig syndrome . Syndrome characterized by b- lipoprotein abnormalities and other alterations. It is manifested by the absence of plasma -lipoproteins, retinitis pigmentosa, acantocytosis of red blood cells, and progressive neurological disorders that begin in childhood and are manifested by ataxia, areph1exia, and severe alterations in proprioceptive sensitivity. The cause is unknown.
Bastian syndrome . Syndrome of the complete section of the spinal cord. It includes an acute, bilateral initial phase, with paraplegia and anesthesia below the level of the lesion, cutaneous and tendon areflexia, urinary retention and fecal incontinence; and a later phase characterized by spastic rigidity and recovery of reflexes and muscle tone. The cause is almost always traumatic.
Bazex syndrome . Paraneoplastic acrokeratosis syndrome. It consists of hyperkeratotic lesions, with an erythematous background, on the pinna, nose, and fingers, accompanied by thickening of the nails and perionixis. There is usually a neoplasm of the upper airways.
Behcet's syndrome . Vasculitic syndrome with ocular and mucosal manifestations. Recurrent picture, of chronic evolution, characterized by episodes of conjunctivitis, uveitis and ulcerations of the cornea and mucosa, buccal and genital. The origin is probably viral and in severe forms it leads to blindness.
Benedikt syndrome . Neurological syndrome of the brainstem with involvement of the third cranial nerve and motor and cerebellar manifestations. It is a picture of ocular motility paralysis on the side of the lesion (III cranial nerve), with frustrated hemiplegia, hernitemblor, hemiataxia and hemiasynergia on the opposite side (lesion of the nucleus, red and of the corticospinal pathway). It is produced by hemorrhages, softening and tumors that affect the peduncular region.
Bernard-Soulier syndrome . Hereditary syndrome related to abnormalities in the coagulation mechanism. It is characterized by a tendency to hemorrhage accompanied by mild thrombocytopenia, giant platelets or atypical morphology, prolonged clotting time, and abnormal prothrombin consumption.
Bemhardt-Roth syndrome . Meralgia paresthetica syndrome. It is manifested by paresthesias and other usually mild sensory disorders on the lateral aspect of one of the thighs. It is due to constriction of the femoroccutaneous nerve at the level of its entry into the fascia lata.
Bertolotti syndrome . Syndrome related to structural abnormalities of the spine. It is a triad made up of scoliosis, sacralization of the fifth lumbar vertebra and sciatica, the first two understood as anomalies: basic and the third as a secondary complication.
Bianchi syndrome . Neurological syndrome due to lesion of the parietal cortex. Sensory aphasia, apraxia and alexia, characterized by the inability of the patient to understand the meaning of the words he hears or reads, as if they corresponded to an unknown language. It is typical of certain lesions, almost always tumorous, of the left parietal lobe.
Bloch-Sulzberger syndrome . Pigment metabolism disorder syndrome with multiple body abnormalities. It is characterized by pigmented, vesicular, and later warty skin lesions, associated with defects in the development of the eyes, bones, and central nervous system. It is hereditary and affects almost exclusively women.
Blount-Barber syndrome . Syndrome related to structural abnormalities of the limbs . lower. Association of osteochondrosis of the upper epiphysis of the tibia, internal rotation of the tibia on the femur and moderate flat feet. It is due to the anomalous development, of an isolated nature, of the proximal extremity of the tibia.
Bogorad syndrome . Syndrome that appears in the recovery period of certain cases of facial paralysis. The distinctive feature is the appearance of unilateral lacrimation, on the side of paralysis, which occurs when the individual ingests food. It is attributed to peculiarities of the nerve regeneration process, during which some of the fibrils destined for the salivary glands would connect with others directed to the lacrimal system.
Bonnet syndrome . Neurological syndrome of the base of the skull. It is manifested by complete ophthalmoplegia, miosis, and motor and sensory disorders of the trigeminal nerve, of insidious onset and progressive advancement. It is produced by infiltrating tumors at the base of the skull.
Bonnier syndrome . Neurological syndrome due to damage to central structures with involvement of cranial nerves III, V, VIII, IX and X. It is manifested by vertiginous state, weakness and drowsiness, neuralgia in the trigeminal territory, ocular motility alterations, instability and hearing loss and other alterations. It is produced by lesions that affect the nucleus, Deiters, the associated vestibular pathways and cranial nerves III, V, VIII, IX and X.
Bradyarrhythmia-tachyarrhythmia syndrome . Syndrome of alternating periods of bradyarrhythmia and tachyarrhythmia. It is characterized by alternating periods of bradyarrhythmia (30 to 50 beats / min.) With dizziness or syncope, which can last from minutes to months, and periods of supraventricular tachyarrhythmia (up to. 200 per minute), which can last from minutes to months and be accompanied by palpitations and other symptoms of paroxysmal supraventricular tachycardia. In the intervals between them there may be a normal sinus rhythm. The cause is unknown.
Brissaud-Marie syndrome . Neurological hysteria syndrome. Hemiespasm of the lip and tongue, without justifying cause, without neurological systematization and in a personality with hysterical features.
Brissaud-Sicard syndrome . Syndrome 'caused by irritative lesions of the lump. Spasm of the facial muscles on the side of the central lesion, with hemiplegia or hemiparesis on the side opposite the lesion.
Bristowe syndrome . Corpus callosum tumor syndrome. It is characterized by progressive hemiplegia on the side of the injury, mild contralateral hemiparesis, dysphagia, stupor, lethargy, coma, and death.
Brown-Séquard syndrome . Spinal cord hemisection syndrome. Flaccid paralysis, muscle atrophy and abolition of deep sensation on the side of the injury, with preservation of superficial sensation, associated with loss of superficial sensation with preservation of motor skills and deep sensation on the side opposite to that of the section. The cause is almost always traumatic (projectiles from firearms).
Bruns syndrome . Neurological syndrome of tumors of the fourth ventricle. It is characterized by intense and intermittent headache, vertigo, vomiting, and visual disturbances, which are accentuated when the patient makes a sudden movement of the head.
Budd-Chiari syndrome . Syndrome of obstruction of the suprahepatic veins. The acute form is characterized by epigastric pain, rapidly progressive painful hepatomegaly, severe ascites, and mild jaundice, and leads to death within days or 1 to 2 months. In the chronic form, the onset is insidious and the manifestations gradually accentuate hepatomegaly with great splenomegaly, abdominal pain, ascites, and edema of the lower limbs. The obstruction is caused by thrombosis (endophlebitis, polycythemia) or extrinsic compression (eg, hypernephroma).
Bulbar syndrome . see Dejerine Syndrome , def 2.
Hernilateral bulbar syndrome . see Babinski-Nageotie Syndrome .
Lateral bulbar syndrome . see Wallenberg syndrome .
Burnett syndrome . see Milk-alkali syndrome .
Bumier syndrome . Endocrine and neurological syndrome of tumor origin. It is characterized by dwarfism, adiposogenital dystrophy, and optic nerve atrophy and is usually caused by slow-growing tumors that compress the pituitary and the chiasm region.
Bywaters syndrome . see Crush syndrome .
Camurati-Engelmann syndrome . Progressive diaphyseal dysplasia syndrome . It is characterized by symmetrical thickening and increased diameter of the diaphysis, of the long bones, with pain in the affected areas, fatigue, abnormal gait and muscle atrophy. The cause is unknown.
Canada-Cronkite syndrome . Intestinal polyposis syndrome with secondary organic alterations. It is characterized by the presence of an intestinal polyposis. multiplex, leading to chronic enteropathy with steatorrhea. As a consequence of this, due to multiple nutrient deficiencies, alopecia, skin changes and nail atrophy occur. The cause is unknown.
Condylar canal syndrome . Neurological syndrome of the base of the skull. Unilateral paralysis of the tongue produced by compression of the XII nerve at the base of the skull, usually of tumor origin
Caplan syndrome . Arthritic syndrome associated with pneumoconiosis. It defines the association of rheumatoid arthritis with the presence of multiple pulmonary nodules (rheumatoid granulomas) in patients with underlying pneumoconiosis. The disorder hinders the transport of gases through the alveolar wall.
Capsulothalamic syndrome . Neurological syndrome due to lesion of subcortical structures. It is characterized by hemianopia, hemianesthesia, partial hemiplegia, alterations in the perception of pain and emotional instability. It is caused by vascular lesions or tumors of the optic thalamus and the internal capsule.
Malignant carcinoid syndrome . Small intestine carcinoid tumor syndrome. It is typically characterized by paroxysmal episodes of cyanotic flushing of the face and neck, abdominal cramps, diarrhea, hypotension, and bronchial spasm; and in more advanced stages due to pigmentary alterations, ascites, endocardial lesions and heart failure. There are usually multiple liver metastases and the condition is due to hypersecretion of serotonin by tumor cells.
Centromedullary syndrome . Neurological syndrome due to cervical cord injury. It is characterized by variable paresis of all four limbs, but disproportionately greater in the upper limbs. It is caused by lesions of the central portion of the cervical cord, of vascular or degenerative origin.
Centroposterior spinal cord syndrome . Neurological syndrome due to spinal cord injury. It is manifested by vasomotor disorders and syringomyelic-type dissociation of sensitivity (loss of thermal and pain sensitivity, with preservation of tactile and deep sensitivity). It is produced by lesions, of the posterior central portion of the gray matter of the spinal cord
Post-traumatic brain syndrome . Residual syndrome far from head trauma. The condition is predominantly subjective polymorphic, but it is frequent in this type of patient and is manifested by headache of variable intensity and location, amnesia, vertigo, tinnitus; palpitations, fatigue, irritability, insomnia and difficulty concentrating.
Cervical syndrome . Pain syndrome related to organic alterations of the cervical spine. It is characterized by periodically presenting neck pain, radiating to the back, shoulder and / or upper limb. It can be symmetric or unilateral, and is usually exacerbated by rotational or extension movements of the head. It is due to compression or irritation of the cervical nerve roots by arthritic impingement.
Cestan syndrome . Neurological syndrome due to extensive damage to brain structures. Hemiplegia and hemianesthesia on the opposite side of the injury, with hemiasynergia, laryngoplegia, lateropulsion and Horner's syndrome on the same side of the injury. The picture is due to scattered lesions that affect the pyramid, the sensory pathways, the inferior cerebellar peduncle, the nucleus ambiguus, and the oculopupillary center.
Scimitar syndrome . Radiological syndrome caused by the anomalous mouth of the pulmonary veins. It is observed in the frontal chest X-rays and consists of a modification of the cardiac image, which becomes convex to the right (due to the opening of the right pulmonary veins in the inferior vena cava) and concave to the left (due to displacement of the heart towards the right lung which is hypotrophic). Overall the image resembles that of a scimitar, in which the blade is represented by the heart and the handle by the great vessels.
Claude Bernard-Horner syndrome . Sympathetic cervical paralysis syndrome. It is characterized by subsidence of the globe (enophthalmia), ptosis of the upper eyelid with slight elevation of the lower eyelid and narrowing of the eyelid fissure, miosis, anhidrosis, and vascular congestion on the affected side of the face. The cause is usually tumor (compression).
Claude-Lhermitte syndrome . Neurological syndrome due to compression of the infundibulum and the third ventricle. It is manifested by a complex, polymorphic picture, which includes narcolepsy or hypersomnia, vasomotor and thermal regulation alterations, diabetes insipidus and a variety of additional symptoms. The cause is generally tumor.
Systolic click-end-systolic murmur syndrome . Mitral valve prolapse syndrome. It is characterized by the auscultation of a late systolic click, in the meso or end-systole, followed by a high-pitched systolic murmur, crescendo-decrescendo, due to regurgitation through one of the mitral leaflets. The patient may have symptoms (episodes of tachycardia, palpitations, undefined chest pain, etc.) or not. It is a usually benign disorder that predominates in young women.
Clouston syndrome . Hydrotic ectodermal dysplasia syndrome. It is characterized by palmoplantar hyperkeratosis, skin hyperpigmentation, hypotrichosis, nail destruction, and often mental retardation. Transmission is autosomal dominant.
Disseminated intravascular coagulation syndrome . Syndrome associated with thrombosis and disseminated hemorrhage at the level of the microcirculation. It is a picture of generalized microthrombosis accompanied by nappa hemorrhages, often irreversible, which is due to the intervention of multiple overlapping mechanisms: consumption and depletion of coagulation factors, generalized damage to the capillary endothelium, exaggerated fibrinolysis, shock, etc. The process is accompanied by thrombocytopenia, fibrinogenpenia, and excess fibrin breakdown products in the bloodstream. It is observed in severe burns and injured persons, in septic shock, in leukemia, and in other conditions of similar gravity.
Cogan syndrome ., Syndrome associated with rapid vision disorders and hearing. Presence of reversible non-syphilitic interstitial keratitis, accompanied by vertigo, tinnitus, nystagmus and rapidly evolving, irreversible deafness. It affects young adults and the cause is unknown.
Cauda equina syndrome . Neurological syndrome due to compression of the lumbar and sacral roots . Flaccid paraplegia, tendon areflexia, painful saddle anesthesia (anal, perineal, genital and gluteal), sphincter disorders and impotence. It is produced by trauma or tumor compression.
Pancreatic cholera syndrome . see WDHA Syndrome .
IBS syndrome . Syndrome of a benign chronic condition of the large intestine. It is characterized by alterations in intestinal motility, with colic and periods of constipation and diarrhea, and usually exaggerated secretion of mucus. It is considered a functional disorder.
Syndrome. of Collet-Sicard . Neurological syndrome of the base of the skull. The picture is identical to that of Villaret syndrome (unilateral lesion of the IX, X, XI, and XII cranial nerves), but without cervical sympathetic paralysis. The: causes are also the same.,
Vertebral artery compression syndrome . see Cervical Vertigo Syndrome
Internal auditory canal syndrome . Sensorineural syndrome of auditory nerve injury. Deafness of perception, corneal hypoesthesia and hyporeflexia or vestibular areflexia. The most frequent cause is the neurinoma of the auditory nerve.
Remnant cystic duct syndrome . see Cystic Stump Syndrome
Narrow Lumbar Duct Syndrome . Peripheral neurological syndrome due to irritation of nerve roots. It is characterized by low back pain accompanied by crural radiculalgia or sciatica, which appears or becomes very severe when walking (intermittent radicular claudication); occasionally there are foot paresthesias, muscular paresis, and discrete sphincter disorders. It is caused by congenital narrowing of the lumbar duct, but it usually manifests itself in general after the age of 50, due to the addition of other pathological processes (osteophytosis, etc.).
Medullary cone syndrome . Neurological syndrome of the lesions of the terminal segment of the cord. Pudendal anesthesia, sometimes in a 'saddle' distribution, urinary and fecal incontinence due to sphincter dysfunction, abolition of ejaculation and erection, and loss of anal and plantar reflexes, and the cause is usually tumor or traumatic.
Acute Pulmonary Heart Syndrome . Acute circulatory insufficiency syndrome of pulmonary origin. It occurs abruptly and manifests as severe dyspnea, cyanosis or lividity, venous hypertension with jugular engorgement and systemic arterial hypotension, dilation of the pulmonary artery, and a shift in the electrical axis of the ECG to the right. Mortality is high and is caused by embolism or thrombosis of the pulmonary artery or its branches.
Chronic Pulmonary Heart Syndrome . Right heart failure syndrome due to prolonged hypertension of the minor circulation in chronic neuropathies. It is characterized by dyspnea on exertion, with or without asthma attacks, chronic cough, clubbing, cyanosis, headache, and drowsiness; in the advanced stages, congestive hepatomegaly and peripheral edema. There is polyglobulia, radiological signs of underlying lung disease, and typical electrocardiographic changes (tall, pointed P wave in II, III, and aVF and negative in aVL, tall R wave in VI, etc.). It is seen in emphysema, chronic bronchitis and bronchiectasis, pulmonary tuberculosis, and other conditions.
Anterior cord syndrome . Neurological syndrome due to spinal cord injury. The lesion is located in the anterior portion of the spinal cord, so there is hypoalgesia, hypoesthesia, and complete paralysis below this level, with relative preservation of the functions that depend on the posterior cord (tactile, vibratory and positional sensitivity). The cause is usually traumatic (firearm projectiles).
Choreic syndrome . Neurological syndrome due to lesion of the gray nuclei of the base. Abnormal, wide, anarchic, involuntary and irrepressible muscular movements, like those of a strange dance, that do not obey any systematization and that disappear during the hours of sleep; it is associated with muscular hypotonia. There is a progressive hereditary chronic form (Huntington's carea) and an acute one that accompanies pregnancy or certain cases of rheumatic fever (Sydenham's carea). It is caused by lesion of the gray nuclei of the base (caudate, putamen, pallidum, center of Luys and locus niger).
Intermediate coronary syndrome . General name of coronary ischemia pictures that do not correspond to a myocardial infarction, nor do they conform to the characteristic pattern of angina pectoris.It includes three different symptoms: a) the syndrome of acute coronary insufficiency, which is characterized by the isolated appearance of an intense painful episode, often at rest, accompanied by electrocardiographic alterations: typical: in the T wave and in the ST segment, but without modifications in the QRS complex; b) the syndrome of unstable angina similar to the previous one but defined by the presentation of repeated episodes during rest, and c) the syndrome of progressive or pre-infarct angina, in which the episodes appear in response to less and less intense efforts; or with more; frequency, or with a longer duration, or with qualitative modification of its characteristics.
Pulmonary emphysema rib syndrome . Pain syndrome associated with chronic obstructive pulmonary disease. Constant pain that the patient refers to the perimeter of the base of the thorax, and that usually intensifies during the usual complications in this condition (acute bronchitis; etc.). It is attributed to structural factors (hyperexpansion of the rib cage, senile osteoporosis, etc.) and the permanent muscular effort that the individual makes to breathe.
Costen syndrome . Temporomasticatory dysfunction syndrome. It is characterized by pain in the temporomandibular region, accompanied by creaking of the corresponding joint, lateralization of the lower jaw when opening the mouth, and occasionally lockjaw; There may also be auditory symptoms (pain, hearing loss, ringing). It is due to malocclusion of the temporomandibular joint, with irritation of the chorda tympani and auriculotemporal nerves.
Cervical rib syndrome . see Syndrome of the superior narrow thorax .
Costoclavicular syndrome . see Syndrome of the superior narrow thorax .
Courvoisier-Terrier syndrome . Vater ampulla carcinoma syndrome. The picture is characterized by jaundice, acholia and coluria, associated with a marked dilation of the gallbladder, but without the rapid cachexia that accompanies carcinoma of the head of the pancreas.
CREST syndrome. Scleroderma associated syndrome. Its name derives from the main manifestations of the picture: C of calcicosis, due to the presence of scattered calcium deposits at the subcutaneous and periarticular level; Raynaud's R, for the frequency with which Raynaud's phenomenon appears in the initial stages of the disease; ES for sclerodactyly, due to the predominance in the fingers of the rigidity that affects the whole body, and T for telangiectasia, due to the development of this type of abnormalities on the skin surface. It is generally seen in the less severe forms of progressive systemic sclerosis.
 Crigler-Najjar syndrome Crigler-Najjar syndrome
|
Crigler-Najjar syndrome . Neurological syndrome related to congenital abnormalities in bilirubin metabolism. It is characterized by the manifestations of the icteric impregnation of the nuclei of the base (kernicterus) (severe neurological disorders), accompanied by high levels of unconjugated bilirubin in the blood. The disease is evident from birth, usually leads to death during the first year of life, and the cause is an inherited deficiency of glucuronyltrasferase,
Cruveilhier-Baumgarten syndrome . Syndrome of congenital hypoplasia of the suprahepatic vein system.The condition is caused by the occlusion of the intrahepatic branches of the indicated veins, and its manifestations, which are serious, derive from direct organic damage caused by the malformation (chronic fibrous liver disease); the resulting hemodynamic repercussion (portal hypertension, with esophageal varices, notable splenomegaly and hypersplenism); of the compensatory function exerted by the umbilical veins, which do not collapse at birth (collateral circulation towards the superior vena cava, soft, continuous, auscultable murmur in the periumbilical region), and of eventual complications (liver failure, digestive bleeding, etc.). Unlike other processes with similar characteristics, it is not accompanied by ascites.
Corpus callosum syndrome see Bristowe syndrome .
Paraneoplastic skin syndrome . Skin syndrome associated with tumor processes in other regions of the body. It includes a variety of conditions that precede or accompany the appearance of the clinical manifestations of a tumor, including acanthosis nigricans (gastric cancer, and less frequently, intestinal, pulmonary or gynecological tumors), dermatomyositis (mammary or genital neoplasms, and sometimes digestive or respiratory), acquired ichthyosis (Hodgkin's disease and malignant hematopoietic conditions), Bazex acrokeratosis (cancer of the larynx), and so on.
 Chotzen syndrome |
Cyriax syndrome . Painful syndrome of the costal cartilages. It is characterized by pain of varying intensity in the parasternal region that relatively frequently radiates to the neck, shoulder, and / or arm. The picture is similar to that of angina pectoris, from which it differs due to its persistent nature and because it is exacerbated when one or more of the costal cartilages is palpated, where the cause of the problem lies.
Charlin syndrome . Nasal nerve neuralgia syndrome. Its features are: 1) intense pain, unilateral, in paroxysmal crises, in a nasoorbital region, 2) ipsilateral hydrorhea, and 3) corneal trophic lesions. It is due to neuritis caused by conditions of the nose.
Chauffard-Still syndrome see Still's syndrome .
Chiari-Frommel syndrome . Non-puerperal galactorrhea syndrome variety. It is typical of the very distant postpartum and is characterized by prolonged lactation, amenorrhea and genital atrophy, in the first case due to sustained prolactin hypersecretion, and in the remaining two cases due to the concomitant decrease in the secretion of pituitary gonadotropins and as a consequence of this, of estrogens by the ovary. The cause is not known precisely, but it is located in the pituitary itself or in the hypothalamus.
Chotzen syndrome . Hereditary syndrome characterized by predominantly cranial and digital malformations . It designates the combination of acrocephaly with polydactyly and partial syndactyly, to which facial malformations (ocular hypertelorism) and mental retardation are sometimes added. The condition is transmitted as an autosomal dominant trait.
Christ-Siemens syndrome . Anhydrotic ectodermal dysplasia syndrome. It is characterized by: a) smooth and lustrous skin, absence of sweat glands and poor hair formation; b) saddle nose and prominent forehead and chin; c) absence of taste and smell; d) dental abnormalities and e) mental retardation. It is transmitted as an X-linked trait.
 Churg-Strauss syndrome |
Churg-Strauss syndrome . Allergic granulomatosis syndrome. Prominent eosinophilia, diffuse pulmonary infiltrates, and association with bronchial asthma. It is a similar vasculitis. to that of polyarteritis nodosa, which also affects the heart, kidney, intestine and peripheral nerves.
Da Costa syndrome . Neurocirculatory asthenia syndrome. Chronic symptom complex 'characterized by suffocation, dizziness, a feeling of fatigue, chest pain and palpitations, without organic cardiac alterations. It is emotional in nature and is also called a soldier's heart.
Dandy-Walker syndrome . Neurological syndrome resulting from a congenital obstacle. in the circulation of the cerebrospinal fluid. It is manifested from birth by intracranial hypertension, hydrocephalus and predominantly cerebellar disorders. The cause is congenital atresia of the foramina of Magendie and Luschka of the fourth ventricle, which prevents normal drainage of cerebrospinal fluid.
Debré-Sémélaigne syndrome . Hypothyroid cretinism syndrome with muscle disorders. Association of hereditary cretinism increased muscle volume, and slow contraction of muscles and tendon reflexes. It is of genetic cause and is transmitted as an autosomal recessive trait.
Nicotinic acid deficiency syndrome . Syndrome of the deficiency of an essential nutrient. In its typical form (pellagra) it is characterized by digestive manifestations (diarrhea, nausea and vomiting), skin: (facial pigmentation, erythema of the base of the neck) and mucous membranes (atrophy of the papillae; lingual, scarlet, rough and fissured tongue), and in severe cases, due to neurological disorders. The disorder is rare, but can be seen in patients with cancer or chronic diarrhea.
Adenosine deaminase deficiency syndrome . Congenital immunodeficiency syndrome with severe osteocartilaginous abnormalities. It is manifested in the first 6 months of life by: a) vomiting, diarrhea and developmental arrest; b) metaphyseal anomalies, cup deformation of the costochondral junctions and short limbs; and c) severe recurrent infections; predominantly respiratory, skin and digestive, caused by viruses, bacteria, fungi or protozoa. Lymphopenia is observed in circulating blood, with a marked decrease in T lymphocytes, and sometimes also in B lymphocytes.
Isolated immunoglobulin M deficiency syndrome . Hereditary immunodeficiency syndrome. It is manifested by low levels of circulating IgM, with atopic and gastrointestinal disorders, splenomegaly and a tendency to the development of severe recurrent infections; and evil processes. The disorder is familial and four times more common in men than in women.
Hepatic aldolase deficiency syndrome . Syndrome produced by a congenital abnormality in fructose metabolism. It is characterized by early arrest of development, a marked tendency to hypoglycemia, elimination of fructose in the urine, kidney disorders, and liver disorders that in severe cases end in cirrhosis with ascites and splenomegaly. The cause is a congenital deficiency of aldolase in the liver.
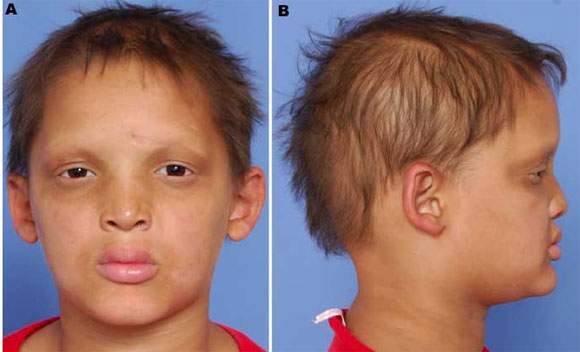 Christ-Siemens syndrome |
Phosphorylase-b-kinase deficiency syndrome . Hereditary syndrome with liver disorders and hypoglycemic manifestations. It is characterized by hepatomegaly, fasting hypoglycemia, and growth retardation. It is a benign condition that is transmitted as an X-linked trait and whose manifestations usually disappear in adolescence.
Iron deficiency syndrome . Syndrome of the deficiency of an essential nutrient. Includes general manifestations (asthenia, fatigue, hypersensitivity to cold, etc.) mucocutaneous alterations; (atrophy of the lingual papillae, angular stomatitis, dry skin, etc.), and mainly paleness and anemia with microcytosis and hypochromia, decreased serum iron, increased ability to combine iron, and absence of this mineral in the extended bone marrow. It is caused by absolute or relative deficiencies (dairy diet, multiple pregnancies), by exaggerated loss (hidden gastrointestinal bleeding, hypermenorrhea) or by a combination of these mechanisms.
Deficiency syndrome purine nucleoside phosphorylase . Congenital immunodeficiency syndrome with hematological and connective tissue abnormalities. The condition does not appear at birth but after the first year of life, mainly due to recurrent infections associated with a progressive decrease in circulating blood T lymphocytes, and sometimes with alterations in supporting tissues and hemolytic anemia. It is hereditary and is transmitted as an autosomal recessive trait.
Vitamin A deficiency syndrome . Syndrome of the deficiency of an essential nutrient. It is characterized by skin changes (keratosis, xeroderma, follicular hyperkeratosis), eye disorders (xerophthalmia, asthenopia, difficulty with night vision) and ease of contracting infections, particularly of the respiratory tract. The condition is rarely seen in its pure form, due to the abundance of vitamin A in food, but it is seen as a component of severe states of malnutrition.
Vitamin B deficiency syndrome . (thiamine). Specific nutrient deficiency syndrome. It gives rise to beriberi and includes general manifestations (asthenia, anorexia, etc.), neuromuscular disorders (weakness of the lower limbs; polyneuritis; with areflexia or patellar hyporeflexia and paresthesias, etc.) and cardiovascular alterations in the wet variant of the condition ( cardiomegaly, congestive heart failure, etc.). It is seen as a component of multicarential symptoms, with some frequency in malnutrition in alcoholics, and occasionally in infants and in pregnancy.
Vitamin B 2 (riboflavin) deficiency syndrome . Syndrome of the deficiency of an essential nutrient. It is manifested by cutaneous alterations (cheilitis, annular stomatitis; sebaceous dysfunction, etc.) and of the mucous membranes (glossitis, atrophy of the lingual papillae), and by ocular disorders (asthenopia, amblyopia, vascularization of the cornea). It is observed, almost always, 'as a component of multi-differential tables.
Vitamin B 6 (pyridoxine) deficiency syndrome . Syndrome of the deficiency of an essential nutrient. Includes mucocutaneous alterations. (nasolabial seborrhea glossitis), neurological disorders (peripheral neuropathy, which predominates in the lower limbs), and in certain cases of childhood, seizures resistant to the usual medication. It is almost always seen as a component of multi-trader frames.
Vitamin B 12 (cyanocobalamin) deficiency syndrome . Syndrome of the deficiency of an essential nutrient. It gives rise to the picture of pernicious anemia, which includes cutaneous and mucosal changes (paleness, mild lemon-yellow jaundice, glossitis), neurological manifestations (paresthesias, ataxia, tendon areflexia, optic nemitis) and hematological abnormalities (hyperchromic anemia, megalocytosis, presence of megaloblasts in bone marrow). It is caused by an insufficient supply of the vitamin or by a lack of gastric intrinsic factor (which is essential for its absorption), either as a consequence of a gastrectomy or of constitutional origin (with acholia and hypochlorhydria).
Vitamin C (ascorbic acid) deficiency syndrome . Syndrome of the deficiency of an essential nutrient. The classic form is scurvy and is manifested by fatigue and emotional disturbances, gingival swelling and bleeding, and muscle pain in adults; and in children due to pain and swelling of the lower limbs, radiological alterations of the bones (subperiosteal hemorrhages, ground-glass appearance, etc.), gingival swelling and bleeding, and bleeding in other regions of the body. It is a currently very rare deficiency, which can affect individuals who live alone and occasionally infants.
Vitamin D deficiency syndrome (cholecalciferol). Syndrome of the deficiency of an essential nutrient. It is manifested, in the child, by the symptomatic complex of rickets (craniotabes, delayed dentition, pelvic and rib cage deformations, abnormal bend of the legs, etc.); and in adults for tetany and osteomalacia that predominates in the skull, pelvis; and knees. The picture is observed in cases of food deficiency (with insufficient endogenous production) of exaggerated loss (malabsorption syndromes) or in rickets resistant to vitamin D (chronic renal failure, renal tubular disorders, etc.).
Vitamin K deficiency syndrome (menadione). Syndrome of the deficiency of an essential nutrient. In the newborn, it manifests as cranial or sternocleidomastoid hematomas, meningeal, oral or umbilical cord hemorrhages, and purpuric lesions; It appears two or three days after birth and is due to the inability to absorb vitamin K due to the absence of normal intestinal flora. In adults, it is evidenced by petechiae, ecchymosis, bruising or hemorrhage, and is generally a consequence of the therapeutic use of coumarin anticoagulants.
Dejean syndrome . Neurophthalmic syndrome due to orbital floor conditions. Includes exophthalmia and diplopia due to displacement of the eyeball; intense pain in the region of the upper jaw, and numbness in the area innervated by the first two branches of the trigeminal (ophthalmic and upper jaw). The cause is usually tumor.
Déjerine syndrome . Neurological syndrome of lesions of the sensory cortex. It is manifested by the loss of a variety of highly specialized functions, such that the patient is unable to recognize a known object by touch (astereognosia), to recognize changes in the position of body segments or differences in the intensity of stimuli, etc. It is caused by vascular lesions (embolism) and sometimes by tumors. // 2. Neurological syndrome associated with focal lesions of the anterior portion of the bulb.On the side opposite to that of the injury, there is hemiplegia that respects the face, while on the side of the injury, paralysis and atrophy of the tongue (XIT nerve) is observed when the injury is superior, or paralysis of the soft palate and cord vowel (X nerve) when located at a level.
Déjerine-Klumpke syndrome . Lower-type brachial palsy syndrome. It is an atrophic paralysis of the muscles of the hand and fingers, associated with Claude's syndrome. Bernard-Homer. It is due to injury to the C8 and DI roots, usually due to compression.
Déjerine-Sottas syndrome . Progressive hypertrophic polyneuropathy syndrome. It manifests itself initially by pain and paresthesia in the feet, and later by weakness and atrophy of the distal segments of the limbs, tendon areflexia, and early disability. The nerves are somewhat thickened by the exaggerated deposit of collagen material. The disorder is inherited and is transmitted as an autosomal recessive trait.
Del Castillo-Trabucco-de la Balze syndrome . Syndrome associated with the congenital absence of the germinative epithelium. It is characterized by sterility and small testicles, with normal libido and secondary sexual characteristics. The testis contains normal Leydig and Sertoli cells, but no germ cells.
Dercum syndrome . Painful adiposis syndrome . Presence of painful, circumscribed fatty deposits in the subcutaneous tissue of the extremities and sometimes in other sectors of the body. It is usually of sporadic appearance, with some documented cases of family incidence.
Cardiovascular deconditioning syndrome . Ergometric syndrome of normal untrained individuals. It manifests itself during stress tests controlled by heart rate and blood pressure that rise normally and very rapidly, without significant electrocardiographic abnormalities and with a mediocre effort capacity. Represents the ideal condition to indicate physical exercises
Decerebrate syndrome . Neurological syndrome due to autonomy of the brainstem. It is characterized by hyperextension of the lower limbs with marked spasticity, fixed flexion of the arms on the chest wall, hyperextension of the head, ocular divergence, and a coma in which only vegetative functions are maintained. It is due to lesions that interrupt nerve conduction at the level of the quadrigeminal tubercles, of traumatic or tumor origin or due to third ventricular hemorrhages.
De Toni-Debré-Fanconi syndrome . see Fanconi syndrome .
Watery diarrhea syndrome . see WDHA Syndrome .
Di Ferrante syndrome . Mucopolysaccharidosis syndrome VIII Form similar to that of Morquio or Sanfilippo syndromes, with stature and mental retardation, discrete hepatomegaly and splenomegaly, hearing loss, and metachromatic granules in leukocytes.
Dighton-Adair syndrome . Osteogenesis imperfecta syndrome. Its basic features are: a) generalized osteoporosis with marked fragility of the bones, and with time the development of multiple bone deformations produced by osteomalacia and the healing of fractures; b) blue sclera, and c) deafness due to osteosclerosis, in about a third of cases. The levels of calcium and phosphorus in the blood and urine are normal. The picture is familiar and is transmitted as an autosomal dominant trait.
Acute stomach dilation syndrome . Complicated postoperative syndrome. Abundant and continuous vomiting, rapid abdominal distention and progressive splashing, and presence of a large fluid level on plain abdominal radiography, performed in a standing position. It occurs in individuals who have undergone intra-abdominal surgery, when they have not undergone gastric aspiration through a tube.
Dysarthria-clumsy hand syndrome . Lacunar neurological syndrome due to injury to the knee and the anterior arm of the internal capsule. It is characterized by dysarthria, facial paralysis and paresis of the tongue, accompanied by slight weakness and clumsiness of hand movements. The cause is vascular (thrombosis, embolism) in the lenticulostriate territory.
Intervertebral disc syndrome . Neuromuscular syndrome due to root compression. It is characterized by usually intense pain in the lumbar region, which appears during or shortly after an intense effort in flexion or hyperextension of the trunk on the lower limbs, and which is characteristically exacerbated by movements and maneuvers: which increase the pressure of the cerebrospinal fluid (cough, defecation, etc.). The pain often radiates to the territory of the sciatic (posterior aspect of the thigh and lateral portions of the leg and foot) and in severe cases is accompanied by hyporeflexia or achilian aref1exia and signs of nerve degeneration on electromyogram. It is due to compression of a nerve root by protrusion of a lumbar intervertebral disc.
Dysphagia and dysphonia syndrome . Neumogastric injury neurological syndrome. It is characterized by unilateral changes including soft palate paralysis, loss of gag reflex, and vocal cord paralysis. It is due to an intracranial lesion, usually of tumor origin.
Placental dysfunction syndrome . Intrauterine fetal distress syndrome. It is characterized by intense yellow pigmentation of the sebaceous daub, the skin and nails of the fetus, greenish pigmentation of the umbilical cord, associated with gigantism and often death of the fetus before birth. It is typical of pregnancies that last beyond three hundred days, and is produced by degeneration of the placental structures.
Temporomasticatory dysfunction syndrome . see Costen syndrome .
Gonadal dysgenesis syndrome . see Turner syndrome
Congenital dyskeratosis syndrome . Hereditary syndrome characterized by skin changes and a tendency to develop malignant processes. It is defined by the presence of reticular hyperpigmentation, palmar and plantar hyperkeratosis, mucosal leukoplakia, and loss of nails; there is also pancytopenia, and a high tendency to develop carcinomas. It is transmitted as a sex-linked character.
Muscular Dystrophy Syndrome Postmenopausal . Myopathic syndrome of unknown etiology It is characterized by progressive weakness of the proximal muscles of the lower and upper limbs, with increasing difficulty in performing normal movements and activities.
Reflex sympathetic dystrophy syndrome . Painful syndrome accompanied by muscular, trophic and vasomotor disorders. Appears in an extremity shortly after a triggering pathological situation (myocardial infarction, trauma, neurological condition) and manifests as distal swelling (eg of a whole hand or a whole foot), severe and burning pain in the same region, and vasomotor lability with episodes of vasoconstriction or vasodilation. It can remain unchanged for days or weeks and then reverse completely, or progressively evolve until producing severe trophic changes, muscle atrophy, and flexion contracture. The cause has not been established with precision, although it is generally attributed a reflex sympathetic origin.
Down syndrome . Genetic syndrome characterized by mental retardation and typical abnormalities. Its basic features are: a) short stature and small skull, particularly in its anteroposterior diameter; b) characteristic facies with small nose, oblique eyes and large protruding tongue; c) short little finger, excessive separation between the first two fingers and toes, and presence of simian palmar folds; d) presence of mild skeletal abnormalities, and quite frequently, cardiovascular malformations, and e) mild to severe mental retardation. It is due to a trisomy of chromium soma 21, of spontaneous appearance and more rarely familial.
Dresbach syndrome . Hereditary elliptocytosis syndrome . It is characterized by the presence of oval or elliptical erythrocytes, with little marked hemolysis, mild splenomegaly, and few clinical manifestations. It is transmitted in an autosomal dominant manner and is due to a congenital defect in the erythrocyte cytoskeleton.
Dressler syndrome . Febrile pain syndrome in certain patients with myocardial infarction. It is manifested by a febrile picture that appears 1 to 6 weeks after a myocardial infarction and is accompanied by retrosternal pain with the characteristics of pleuropericardial pain: it radiates to the neck and both shoulders, relieves when the individual leans the torso forward, and it is exacerbated by deep inspiration. The cause is pleuropericarditis, often associated with pneumonitis, and is considered an autoimmune reaction.
Duane syndrome . Congenital syndrome characterized by unilateral eye disorders. It is manifested by the impossibility or marked difficulty of external rotation of the eye, difficulty of internal rotation, retraction of the eyeball and defective convergence. The disorder is hereditary and is transmitted as an autosomal dominant trait.
Duchenne syndrome . Neurological syndrome of progressive bulbar palsy. It is a progressive picture of dysarthria, dysphonia and paralysis, preceded by spasm of the muscles of the lips and tongue, due to the increasing involvement of the V, VII cranial nerves; IX, X and Xll. The condition is degenerative in nature and its cause is unknown.
Dumping syndrome . see Rapid emptying syndrome .
Duplay syndrome . Calcific tendinitis syndrome. It consists of pain and limitation of movement of the shoulder joint, due to inflammation and calcification of the subacromial or subdeltoid bursa.
Eaton-Lambert syndrome . Myasthenic syndrome associated with certain lung neoplasms. It is manifested by weakness of the proximal portions of the upper and lower limbs, accompanied by pain, numbness, and tendon areflexia or hyporeflexia. Unlike the myasthenia gravis syndrome, a) there are no ocular musculature alterations, and b) the force tends to increase, and not to decrease, with the repetition of muscle movements. In general, the condition precedes the appearance of a lung carcinoma by months or up to two years. cells, small.
Eclamptic syndrome . Hypertensive edematous syndrome of pregnancy. It is manifested by the triad of hypertension, edema and albuminuria, in addition to nausea and vomiting, and eventually manifestations of cardiac overload and neurological signs of intracranial hypertension, including seizures and coma. It is typical of the last stages of pregnancy.
Eddowes syndrome . see Digihton-Adair Syndrome
Ehlers-Danlos syndrome . Syndrome associated with hereditary disorders of the connective tissue. It is characterized by hyperelasticity and fragility of the skin, joint laxity, and the facility to develop subcutaneous hemorrhages and muscle hematomas that later calcify. The disorder is transmitted as an autosomal dominant trait.
Eisenmenger syndrome . Syndrome characterized by cardiac malformations and pulmonary hypertension. It is defined by the presence of a high VSD, with thrust of the aorta over the septal defect, hypertrophy of the right ventricle, and dilation of the pulmonary artery, which is evidenced by mid-arch enlargement on frontal chest X-rays.
Sinus node disease syndrome Sinus node dysfunction syndrome of unknown etiology. Clinically, it is manifested by temporary bouts of weakness, easy fatigue, dizziness and sometimes syncope states, which recur with a variable frequency in individuals without signs of underlying heart disease, and which are due to sudden episodes of bradyarrhythmia (sinus bradycardia, sinoatrial block, sinus arrest ). The baseline electrocardiogram is usually normal, so the diagnosis can only be made accidentally (occurrence of the abnormality during recording) or if the clinician suspects it and requests a prolonged continuous Holter recording.
Erb syndrome . see Myasthenia gravis syndrome .
Anterior scalene syndrome . see Syndrome of the superior narrow thorax .
Exertion syndrome . see Da Costa Syndrome .
Posteroexternal space syndrome of the condyle . see Collet-Sicard syndrome .
Retroparotid space syndrome . see Villaret Syndrome
Schizophrenic syndrome . Psychotic syndrome characterized by a profound detachment of the individual from reality, based on severe alterations in personality, thought and affectivity.It is observed in adolescence and young age and is manifested, basically, by: a) personality disorders, with loss of the sense of individuality and uniqueness, and often with the conviction of being controlled by people or forces, alien to himself; b) thought alterations, by which the individual explains his environment on the basis of inconsequential facts of that reality, to which he assigns an arbitrary meaning, and of the rigidly structured rational constructions that he makes from those facts; c) obscuring the forms of expression, which make them cryptic and only comprehensible to himself; d) apathy, inertia, negativism, stupor, and eventually, catatonia and autism; e) pronounced disaffection and f) preservation of intelligence.
Syndrome of the superior stricture of the thorax . Neurovascular syndrome due to irritation of the brachial plexus.It is a symptomatic complex referred to the upper limb or to any of its parts, especially the hand, which is manifested by pain, paresthesia and fatigue; by vasomotor alterations: local (acrocyanosis, paleness, coldness, Raynaud's phenomenon, and occasionally mild edema) and eventually by trophic alterations (digital ulcerations) and localized muscle atrophies. It is due to irritation, compression or stretching of the components of the brachial plexus by neighboring structures, and encompasses conditions of different origin but with similar consequences: a) the anterior scalene syndrome, in which the subclavian artery and the brachial plexus are compressed between the anterior scalene, the first rib and the median scalene; b) cervical rib syndrome,
Malignant exophthalmia syndrome . Thyrotoxicosis ophthalmopathic syndrome. It is manifested by a pronounced bilateral exophthalmia, with paresis of the external oculomotor muscles, swelling of the eyelids, edema and injection of the conjunctiva, and constant retroocular pain. It is typical of Graves' disease.
Fanconi syndrome . General name of the set of anomalies that result from a dysfunction of the proximal tubules. It is observed as an idiopathic form or associated with a variety of pathological processes (amyloidosis, nephrosis, intoxication by expired tetracyclines, etc.), and it is defined based on the following criteria: a) by the presence of four basic anomalies: glucosuria not associated with hyperglycemia, phosphaturia, aminoaciduria, and renal tubular acidosis; b) due to the clinical manifestations of these anomalies that in general can be managed satisfactorily, loss of glucose, formation only of cystine stones, osteoporosis, metabolic acidosis, potassium depletion. etc. and c) by the absence of an associated cystinosis.
Favre-Racouchout syndrome . Congenital syndrome characterized by focal connective tissue abnormalities. It is manifested by the presence of comedones and circumscribed, thickened yellowish plaques that are located on the face, around the eyes and nose.
Felty syndrome . Rheumatoid arthritis syndrome associated with splenomegaly and hematological disorders. It is seen in patients with long-standing rheumatoid arthritis and includes splenomegaly with leukopenia, anemia, and thrombocytopenia, and occasionally, vasculitic phenomena that cause skin ulcers and peripheral neuropathy. The causal mechanism of these alterations is not yet known.
Adrenal feminization syndrome . Variety of adrenogenital syndrome. It is manifested in man by gynecomastia, testicular atrophy, feminoid changes in body conformation and hair implantation, and increased urinary excretion of estrogenic metabolites. The most common cause is an androstenedione-secreting adrenal tumor, which is peripherally converted to estrone and estradiol, or rarely, a deficiency of 3-b-ol-dehydrogenase, which shunts the metabolism of androstenedione to that of progestogens.
Testicular feminization syndrome . Pseudohermaphroditism syndrome in genetically male individuals. It is characterized by the presence of testes, total feminization of the external genitalia and body conformation, and absence or marked hypoplasia of the uterus and tubes, with normal levels of circulating testosterone. They are due to a resistance to testosterone in the effector organs.
Fetal alcohol syndrome . see Fetal alcohol syndrome
Fitz-Hugh-Curtis syndrome Perihepatitis syndrome due to different abdominopelvic infections. It is manifested by fever, pain and contracture of the abdominal wall muscles, more intense pain in the right upper quadrant, and occasionally auscultable friction noise at the level of the right upper quadrant. The most cause; common, it is gonorrhea; although it has also been observed in chlamydial salpingitis and perhaps other causative agents.
Splenic flexure syndrome . Seu doanginous syndrome of digestive origin. It includes pain and discomfort, nonspecific in the left upper quadrant of the abdomen, which can cause pain in the precordial region and in the left shoulder and arm. Its usual cause is colonic meteorism.
Foix syndrome . Neurological syndrome characterized by involvement of cranial nerves V, VII, IX, X and XII, Y due to profrusion of an eyeball. It is manifested by complete unilateral ophthalmoplegia (m, IV and VI cranial nerves) associated with anesthesia of the cornea (ophthalmic branch of the V nerve), with the addition of ipsilateral exophthalmos, which distinguishes it from Rochon-Duvigneau syndrome. It is produced by tumors and aneurysms of the cavernous sinus, or by invasive tumors of the venous sinuses or the sella turcica.
Foix-Alajouanine syndrome . Subacute necrotizing myelitis neurological syndrome. It manifests as a painful paraplegia that is first spastic and then flaccid, with muscle atrophy, tendon areflexia, anesthesia, and sphincter incontinence. The cause appears to be degenerative, secondary to infectious processes or of vascular origin.
Forbes-Albright syndrome . Endocrine syndrome due to prolactin hypersecretion. It is characterized by sustained galactorrhea in women who are not in the puerperal period, accompanied by elevated levels of circulating prolactin. It is caused by tumors of the adenohypophysis and, less commonly, the hypothalamus.
Foster-Kennedy syndrome . Unilateral anosmia syndrome of organic cause. It manifests only as unilateral anosmia and is caused by arterial aneurysms or, more often, by tumors of the base of the frontal lobe that compress the bulb and olfactory band.
Frey's syndrome . Irritative syndrome of certain parotid conditions. It is characterized by redness, sweating and a sensation of heat in the territory of the auriculotemporal nerve, triggered by chewing. It is observed in unilateral lesions of the parotid (tumors, suppurations) or of the cervical sympathetic.
Froin's syndrome . Syndrome related to non-infectious cerebrospinal fluid disorders. It defines the cerebrospinal fluid that is xanthochromic (light yellow, transparent), with high protein content, rapid coagulation and normal number of cells. These characteristics are observed in cases of interrupted flow between the cerebral ventricles and the most distal areas.
Drug-related galactorrhea syndrome . Endocrine syndrome due to prolactin hypersecretion. It is characterized by galactorrhea in women, and by mamillary turgor or gynecomastia in men, which appear during treatment with certain medications (including spironolactone, phenothiazine and other tranquilizers and antidepressants, reserpine, methyldopa, digitalis, etc. .) and that disappear when their administration is interrupted.
Geniculate ganglion syndrome . see Ramsay-Hunt Syndrome
Ganser syndrome Syndrome of certain behavioral disorders in patients with hysteria. It manifests itself in a hysterical personality, by amnesia, atypical disturbances of consciousness, absurd acts, and meaningless answers to questions.
Garcin syndrome . Neurological syndrome of the base of the skull. Unilateral paralysis of all cranial nerves, especially the last ten, without signs of intracranial hypertension. It is caused by extensive tumors or tumor metastases that progressively infiltrate the base of the skull.
Gardner syndrome . Hereditary syndrome characterized by skin changes and a tendency to develop malignant processes. It is manifested by the presence of dermoid tumors, lipomas, fibromas, osteomas, and epidermal and sebaceous cysts, associated with colonic polyposis. The incidence of adenocarcinomas of the colon is extremely high, unless the individual is colectomized. The picture is transmitted as an autosomal dominant trait.
Gastrocardiac syndrome . Functional heart syndrome of gastric origin. It is manifested by extrasystoles, transitory episodes of tachycardia or pseudo-anginal symptoms, unrelated to exertion and that usually appear in the postprandial period. They are attributed to compression of the gastric chamber against the lower wall of the diaphragm and with some frequency there is an associated hiatal hernia.
Gélineau syndrome . Narcolepsy syndrome. In its full form it is characterized by the following tetrad: a) recurrent, irresistible attacks of daytime sleepiness (narcolepsy); b) brief, sudden episodes of loss of muscle tone without loss of consciousness (cataplexy); c) frightening sensation of inability to move voluntary muscles at the beginning of sleep or on awakening (sleep paralysis), and d) hallucinations at the beginning of sleep (hypnagogic) or upon awakening (hypnopompic) The cause has not yet been clarified.
General adaptation syndrome . Syndrome of the organism's global response to stressful situations. Set of systemic reactions consecutive to sustained exposure to stress conditions (trauma, cold, heat, fatigue, etc.). It comprises three stages: alarm reaction, resistance stage and exhaustion stage.
Gerstrnann syndrome . Neurological syndrome due to dominant angular gyrus injury. Combination of tactile agnosia, difficulty distinguishing between right and left, agrafia, acalculia, and often apraxia of grasp. It is seen in lesions of the left angular gyrus, between the parietal and occipital cortex, and is usually of tumor origin.
Gilles de la Tourette syndrome . Chronic multiple tic syndrome. They are characterized by facial tics that appear in childhood and progress to generate jerks: wide; stereotyped, in the rest of the organism, and that are typically accompanied by echolalia and coprolalia. The cause is attributed to a combination of genetic and environmental factors, and the disorder persists throughout life.
Goodpasture syndrome . Syndrome associated glomerulonephritis and pulmonary disorders hemop consumption. The starting point is a respiratory condition with hemoptysis and diffuse, bilateral pulmonary infiltrates, followed by anemia and rapidly progressive glomerulonephritis manifested by hematuria, severe proteinuria, arterial hypertension, and renal failure. The outcome is usually fatal and is attributed to anthoimmunity phenomena.
Gorlin-Goltz syndrome . Hereditary syndrome characterized by skin changes and a tendency to develop malignant processes. It is defined by the presence of multiple basal cell carcinomas, epidermoid cysts, and dimples on the palms and soles; in addition to ovarian fibromas and maxillary cysts, and in a certain proportion of cases, hypertelorism and metacarpal abnormalities. It is transmitted as an autosomal dominant trait and a definite tendency to the development of fibrosarcomas of the lower jaw and medulloblastomas has been demonstrated.
Gougerot-Carteaud syndrome . Confluent and reticulated papillomatosis syndrome. It is characterized by the presence of a large number of discrete papules; which then converge to become warty papules. It is located in the midline of the trunk and flexion folds of the elbow and regresses slowly. It usually affects girls of pubertal age.
Gradenlgo syndrome . Neurological syndrome with involvement of cranial nerves V and VI. It is defined by the presence of unilateral paralysis of the outward gaze (lesion of the sixth cranial nerve) accompanied by headache and ipsilateral facial neuralgia (lesion of the fifth cranial nerve). It is observed in suppurative processes of the ear, middle and in tumors of the crag fossa.
Groenblad-Straudberg syndrome . Elastic pseudoxanthoma syndrome. Its distinctive features are: a) on the skin, the appearance of degenerative rhomboidal areas, atrophy and flaccidity, which predominate in the neck, armpit and groin and develop after 30-40 years; b) progressive decrease in vision, related to the presence of angioid bands, of a brown or grayish-brown color on the surface of the reticulum, and c) increasing signs of peripheral circulatory insufficiency produced by calcification and occlusion of medium-caliber arteries, including those of the upper limb. The condition is of unknown cause and is transmitted as an autosomal recessive trait.
Guillaln-Barré syndrome . Acute idiopathic polyneuritis syndrome: Rapidly progressive weakening of ascending motor neurons that usually occurs after a respiratory or enteric infection. It begins with paresthesias of the feet, followed by flaccid paralysis and weakness of the legs; that ascend to the arms, trunk and face; it is accompanied by low fever, bulbar paralysis, absence or decreased tendon reflexes, and increased cerebrospinal fluid proteins without concomitant cell growth. The cause is thought to be immunological and recovery is complete in 75% of patients.
|
|
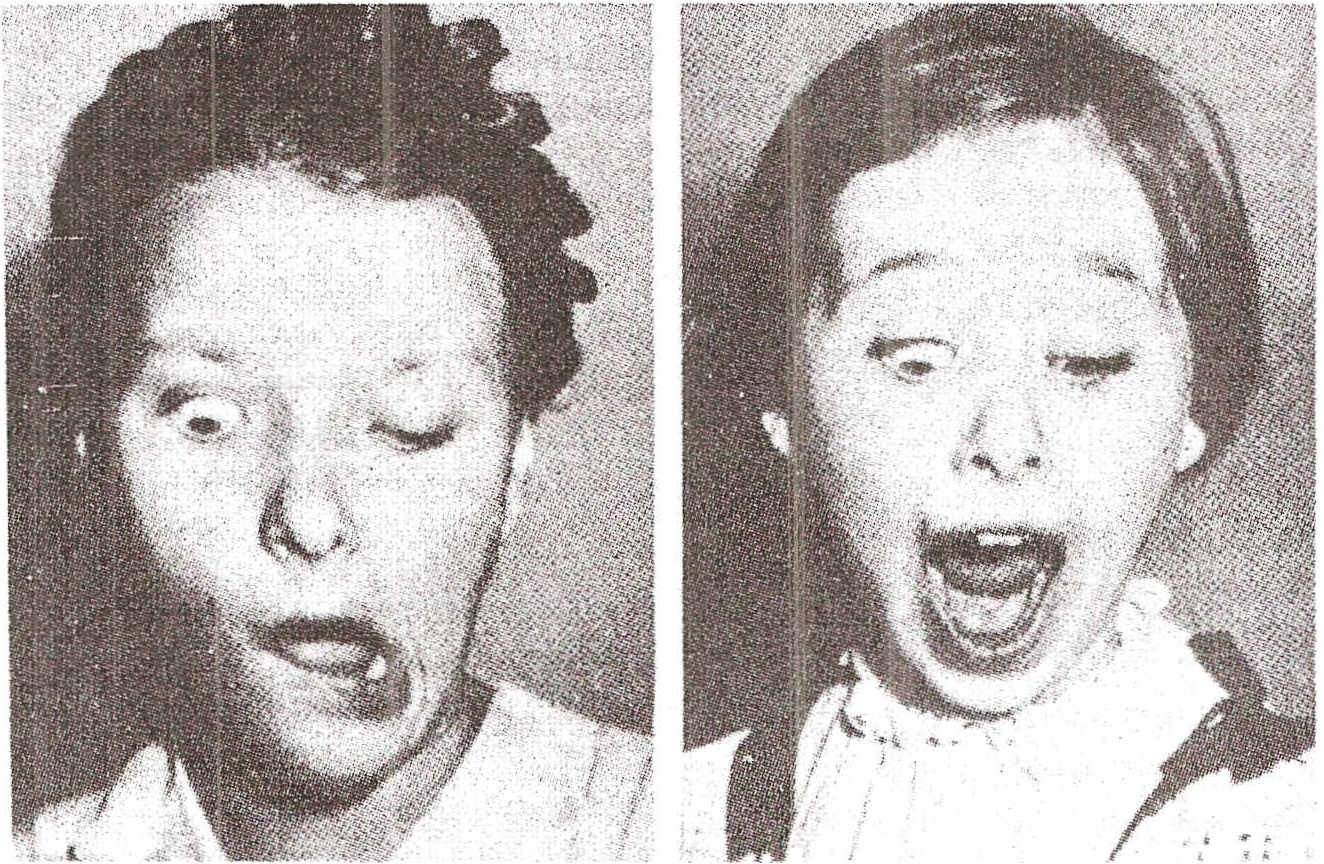
Gunn syndrome . Syndrome related to associated movements of an eyelid and the lower jaw. It is characterized by unilateral eyelid ptosis, almost always congenital, and exaggerated elevation of the ptosus eyelid when moving the mandible in a contralateral direction; In some cases the reverse also occurs: stimulation of the cornea causes movements of the jaw to the opposite side. The cause is unknown and there are no other bodily disorders.
Hallopeau-Siemens syndrome . Polydysplastic bullous epidermolysis syndrome. It consists of blisters that develop from early in life and that heal leaving scars, on which epitheliomas may appear. The mucous membranes are frequently affected. It is a recessive inherited disorder.
Multiple hamartoma syndrome . Hereditary syndrome characterized by skin changes and a tendency to develop malignant processes. It is manifested by the presence of: a) warty papules on the skin; b) fibromas in the oral mucosa; c) multiple hamartomas in other regions of the body (mammary fibrocystosis, neuromas, thyroid adenomas, lipomas and hemangiomas), and d) increased tendency to develop carcinomas. mammary and thyroid gland. The disorder is transmitted as an autosomal dominant trait.
Hamman-Rich syndrome . Idiopathic pulmonary fibrosis syndrome. It is characterized by the development of fibrous tissue in the lung interstitium, of chronic, subacute or acute evolution. There is progressive and later permanent dyspnea, clubbing of the finger, and alteration in the transport of gases through the alveolar wall. It is irreversible and of unknown cause.
Harris syndrome . Endogenous hyperinsulinism syndrome. It is manifested by more or less frequent episodes of restlessness, paleness, sweating, tachycardia, mental confusion and vision disturbances, related to a marked decrease in plasma glucose levels. It is produced by hypersecretion of insulin from tumors or diffuse hyperplasia of the beta cells of the islets of Langerhans.
Hedblom syndrome . Acute inflammation of the diaphragm syndrome. It is manifested by inspiratory pain, immobility of the lower thorax during inspiration, and bilateral pain in the upper abdomen, with no abdominal pathology to justify the condition. It is due to a primary acute myositis of the diaphragm.
Heerfordt syndrome . Uveoparotid fever syndrome. It is characterized by uveal inflammation and parotid swelling, associated with bilateral facial paralysis (facial diplegia) with or without paralysis of other cranial nerves. It is one of the initiation variants of sarcoidosis.
Heidenhain syndrome . Neurological syndrome of a presenile dementia. It is a rapidly evolving insane condition accompanied by cortical blindness, dysarthria, ataxia, athetosis and generalized rigidity, without alterations in the fundus of the eye. The disorder is degenerative and of unknown cause.
Hemangioma syndrome with thrombocytopenia . see Kasabach-Merritt syndrome .
Subdural hematoma syndrome . Neurological syndrome of intracranial venous collections. It is manifested by headache, drowsiness, mental confusion, focal motor signs (monoparesis, hemiparesis), pupillary asymmetry, papilledema, and eventually stupor, coma, and conical seizures. It is usually of insidious progression, overlapping, and is produced by the slow expansion of an intracranial hematoma of venous origin, in patients with or without a striking history of head trauma, of short or medium duration (from a few days to one or more months )
Bitemporal hemianopia syndrome . Neuro-ophthalmologic syndrome of bilateral optic nerve injury. The condition is caused by adenomas of the pituitary gland, meningiomas of the sella turcica or saccular aneurysms of the polygon of Willis, which when expanding simultaneously compress the internal portion of both optic nerves, that is, the fibers that go to the nasal half of both retinas. As a consequence of this, there is blindness to objects located on the temporal side of both visual fields (bitemporal hemianopia).
Sphenoid cleft syndrome . see Rochon-Divigneau syndrome.
Hyperabduction syndrome . see Syndrome of the superior narrow thorax .
Hyperkalemia syndrome . Muscle and electrocardiographic syndrome of hyperkalemia. It becomes evident when serum potassium levels exceed 6 mEq / L, and is characterized by increasing muscle weakness, hyporeflexia or osteotendinous areflexia, and typical changes in the electrocardiogram: first, sharp T waves appear in all leads and then, successively, PR elongation, disappearance of the P wave, alterations in the QRS complex, and eventually ventricular fibrillation and cardiac arrest. It is observed in Addison's disease, in the terminal stages of chronic kidney failure, and mainly in forms. hypercatabolic of acute renal failure.
Palmoplantar hyperkeratosis syndrome . Hereditary syndrome with skin changes and a tendency to develop malignant processes. It is characterized by the presence of hyperkeratosis of the palms of the hands and the soles of the feet, which begins already in second childhood, without other abnormalities: detectable. The subsequent incidence of carcinomas of the esophagus is extremely high, and the condition is transmitted as an autosomal dominant trait.
Hyperkinetic syndrome of infancy . Syndrome of acceleration of the psychomotor functions in the child. It is characterized by motor hyperactivity, impulsivity, restlessness, marked inability to fixate attention, and low tolerance for frustration. The cause is not known, although in most cases it is not organic, and the condition usually disappears in adolescence.
Hypersomnia syndrome with nocturnal apnea . Apneic syndrome associated with obesity of different etiology. The typical picture is that of a daytime hypersomnia with pauses; of abnormal frequency and duration that occur during sleep. It is seen in Pickwick syndrome, simple obesity, myxedema, and other disease states.
Portal hypertension syndrome . Venous hypertension syndrome due to portal system obstruction. It is manifested by ascites and peripheral edema, congestive splenomegaly, development of collateral venous circulation (abdomen and anterior chest wall), dilation of other venous systems (esophageal varices), oliguria, and eventually digestive hemorrhages (ruptured esophageal varices). It is seen as a primary manifestation in portal vein pylephlebitis, and secondarily in liver cirrhosis and other obstructive processes.
Hyperventilation syndrome . Non-organic pulmonary hyperventilation syndrome. It is a picture of anguish and a feeling of shortness of breath, numbness and paresthesia in the face and limbs, and cramps in the hands and feet. It usually occurs during the night, when the individual tries to fall asleep or shortly after falling asleep. The cause is hyperventilation triggered by emotional factors.
Hypokalemia syndrome . Muscular and electrocardiographic syndrome of hypokalemia. It is characterized by general weakness that can even lead to complete paralysis. (as seen in familial periodic paralysis), accompanied by typical electrocardiographic abnormalities: generalized flattening of the waves: T, prominent U waves, ST lowering, and eventually cardiac arrest in asystole. The most frequent cause is the ingestion of diuretics, by self-medication or by medical prescription and it can also be caused by chronic abuse of laxatives, in the pictures of profuse vomiting or profuse diarrhea, and so on.
Intracranial hypotension syndrome . Neurological syndrome of hypotension of the cerebrospinal fluid. It is manifested by nausea, vertigo and tinnitus, bradycardia, headache that relieves in decubitus, neck stiffness, and mild to moderate bilateral papilledema. It is a relatively frequent complication of head trauma and is due to an interruption in the formation of cerebrospinal fluid with the consequent tendency to collapse of the ventricular spaces.
Idiopathic orthostatic hypotension syndrome . Syndrome associated with peripheral or central degeneration of structures of the autonomic nervous system. It is characterized by orthostatic hypotension whose severity can be such as to cause syncope or seizures. The condition is progressive, it is observed in the elderly and is accompanied by ascending anhidrosis, decreased basal metabolism and norepinephrine production, poor tear and saliva secretion, bladder atony, and other manifestations.
Idiopathic hypoventilation syndrome . Hypoventilation syndrome with clinical manifestations, voluntarily correctable, and of unknown cause.It is a rare condition that affects individuals without pulmonary or chest wall pathology, without alterations in respiratory function tests, and without detectable abnormalities in the central nervous system. It is characterized: a) by a sustained involuntary and insensitive tendency to hypoventilate, sufficiently important for the PC02 to rise and the blood P02 to decrease, and for the individual to be in a state of permanent weakness, tiredness and drowsiness, and b) by the individual's ability to reverse the situation to the extent that he or she is aware of what is happening to him, and voluntarily intensifies ventilatory activity. Hematocrit values are greater than 50%, and periodic breathing episodes are observed during sleep in most cases.
Hippel-Landau syndrome . Neuro-ophthalmological syndrome associated with congenital vascular malformations. Anatomically, it is defined by the simultaneous presence of multiple capillary hemangiomas in the retina and one or more hemangioblastomas in the cerebellum, slow growing and with cystic formations. And clinically, due to headache and papilledema, and progressive manifestations of cerebellar ataxia and loss of vision. There are cases of spontaneous appearance and others that are transmitted as an autosomal dominant trait.
Holt-Oram syndrome . Syndrome associated with cardiovascular and skeletal system malformations. Association of interatrial septum defects, or other cardiac anomalies, with hypoplasia of the clavicles and malformations of the upper limbs.
Holzknecht-Jacobson syndrome . Radiological syndrome of pulmonary atelectasis. It is characterized by opacity of the affected lung field, deviation of the mediastinum towards that side; elevation of the ipsilateral diaphragm and retraction of the intercostal spaces.
Stiff man syndrome . see Wolt Man-Moersch Syndrome
Shoulder-hand syndrome . see Reflex Sympathetic Dystrophy Syndrome .
Homen syndrome . Neurological syndrome of the degeneration of the lenticular nucleus. It is a progressive and irreversible condition characterized by vertigo, dysarthria, ataxia, rigidity that predominates in the lower limbs, and dementia. The origin is genetic.
Horner syndrome . see Claude Bernard Horner Syndrome .
Horton syndrome . Histamine headache syndrome It is characterized by unilateral attacks of pain over the eye and forehead, with sudden onset and termination; there is redness, heat, and sweating on the affected side due to dilation of the temporal artery. It predominates in men, during sleep, and is attributed to a local histamine discharge
Hunt syndrome . Juvenile agitant paralysis syndrome. It manifests itself from the first years of life by muscular hypertonia and facial features and general attitude typical of Parkinson's disease. It is familiar and is due to the progressive degeneration of the globe pallidus and other neighboring nuclei.
Hunter syndrome . Mucopolysaccharidosis syndrome II. It is characterized by: a) scaphocephaly with a short neck, hirsute eyebrows, large tongue, and lateral prominence of the outer walls of the orbits; b) joint stiffness, with hollow feet and hands in flexion; c) absence of corneal opacities, and d) deafness, but with less severe mental retardation than that of Hurler syndrome. It is observed only in males, who may reach the middle age of life, and is due to a deficiency of iduronate sulfatase, transmitted as an X-linked recessive trait.
Hurler syndrome . 1H mucopolysaccharidosis syndrome. Its basic features are: a) voluminous head with scaphocephaly, hirsute eyebrows; prominent forehead and eyes, thick tongue and bulging cheeks (gargoylism, due to its similarity to gargoyles in Gothic architecture); b) short stature, neck and limbs, with marked kyphosis and a prominent belly; c) corneal opacities, and d) deafness and severe mental retardation. The disorder is genetic, due to an aL-iduronidase deficiency and death usually occurs before the age of 10
Pigmentary incontinence syndrome . see Dloch-Sijlzberger syndrome .
Severe combined immunodeficiency syndrome . Congenital syndrome related to a global insufficiency of the immune mechanisms. It is characterized by severe disorders of humoral and cellular immunity, with immunoglobulin deficiency and marked lymphopenia including T, and B cells. There are repeated infections and patients usually die within the first year of life. The disorder is of the congenital hereditary type or appears in the form of sporadic cases.
Immunodeficiency syndrome with thymoma . Immune deficiency syndrome of undetermined cause It is observed in patients with spindle cell thymomas and is characterized by the absence or marked decrease of B lymphocytes in the circulation and of their precursors in the bone marrow, together with eosinopenia, low levels of plasma immunoglobulins, and occasionally, erythroid aplasia.
Polyglandular insufficiency syndrome . Syndrome of simultaneous insufficiency of two or more endocrine systems. The syndrome is produced by different endocrine hypofunction pictures (adrenal insufficiency, hypoparathyroidism, lymphocytic thyroiditis, gonadal insufficiency) and by the manifestations that result from the combination of two or more of them with each other, to which diabetes and diabetes are often associated dwarfism. No significant abnormalities are detected in the adenohypophysis and its basic features are low levels of circulating hormones and the existence of antibodies against two or more endocrine systems.
Newborn respiratory distress syndrome . Acute respiratory distress syndrome of multiple etiology. Its two main signs are dyspnea and marked tachypnea (more than 60 breaths per minute), accompanied by variable degrees of tachycardia, cyanosis, and alterations in the acid-base state. It is produced by causes: central (drug depression, cerebral immaturity or anoxia, etc.) or peripheral: infectious processes, aspiration of amniotic fluid, atelectasis, pulmonary immaturity (Wilson-Mikity syndrome), deposit of hyaline material in the alveoli and bronchioles ( hyaline membrane syndrome), etcetera.
Interolivar syndrome . see Dejerine syndrome .
Fructose intolerance syndrome . see Hepatic Aldolase Deficiency Syndrome .
Digitalis intoxication syndrome . Syndrome produced by the ingestion of excessive doses of digitalis. It includes digestive disorders, which are usually the first to appear (marked anorexia, persistent nauseous state), visual disturbances (blurred or colored vision, scotomas) and electrocardiographic abnormalities whose severity is usually proportional to the intensity of the intoxication, arrhythmias of the type appear. Isolated extrasystole, extrasystolic bigeminy, or multifocal ventricular extrasystole, followed by first or second degree atrioventricular block, and eventually, complete block.
Radiation syndrome . Syndrome produced by acute overexposure to roentgen rays. It is manifested by anorexia, weakness and fever, intense thirst and prostration, followed after a short time by anemia, granulocytopenia, weight loss, hair loss, and generalized alterations that in cases of massive irradiation (radioactive leaks, nuclear explosions, etc.) quickly lead to the death of the individual.
Ivemark syndrome . Syndrome of spleen agenesis with cardiac and visceral abnormalities. It is characterized by agenesis of the spleen, cardiac malformations: usually with right-to-left shunting, and inversion of the viscera of variable degree. The picture is congenital and is due to developmental alterations that occur around the fourth or fifth week of intrauterine life.
 Jaccoud syndrome Jaccoud syndrome
|
Jaccoud syndrome . Yes nd r or m and a poliartr i t i s c ró nica pro g resi is d e e v or lu c tio n to you pica .It defines a progressive chronic polyarthritis with consequences virtually comparable to those of rheumatoid arthritis (periarticular fibrosis, stiffness, ulnar deviation of the hand, etc.) but that does not begin as such, but rather as an evolutionary variant in an individual who was a carrier, initially, of the typical reversible manifestations of acute polyarticular rheumatism (rheumatic fever).
Jackson syndrome . Yes n d ro m e neurol or logical p or r l e sion of the trunk c and rebra l c or n co mprom iso of n and rv i or s cranial I X and X is manifested by paralysis half of the tongue on the side of the injury (IX nerve) plus the signs of Avellis syndrome. It is caused by tumors or softening foci at the level of the roof of the bulb and the corticospinal tract.
Jacod syndrome . S iNDr or m e ne urooftálmico po r l and whether or n cran nerve and to the is. It is unilateral and is characterized by neuralgia in the trigeminal territory, blindness, and complete ophthalmoplegia. It is produced by nasopharyngeal tumors that extend to the petro-sphenoidal junction and affect cranial nerves II, III, V, and VI.
Jaddasohn-Lewandowsky syndrome . Sind r or m and d and the cong pachyonychia é n i ta. It is characterized by thickening of the nails; onychogryposis, hyperkeratosis of the knees, elbows, palms and soles, oral leukoplakia and hyperhidrosis of the hands and feet. It is inherited as an autosomal dominant trait.
Jackson-Wartenhorst syndrome . Yes No drome d e l a p or licondritis recidi v ante. It is a feverish, painful inflammatory process of the cartilage of the ears and nose, which leads to atrophy and can affect the cartilage of the respiratory tract and ribs. It is accompanied by arthritis, episcleritis, iritis, myocarditis, and aortic insufficiency. The cause is unknown.
Jervell and Lange-Nielsen syndrome . see QT syndrome .
Kartagener syndrome . S í n d Rome d and one Cua d r or m to lformacione s cardi ov a s c ular is and d and l as pathways resp go to to r ias . It is defined by the presence of dextrocardia, situs inversus, and bronchiectasis and by a tendency to develop recurrent pulmonary and sinus infections. The disorder is inherited and is transmitted as an autosomal dominant trait.
Kasabach-Merritt syndrome . Yes n thrombocytopenic drome related to the presence of a hemangioma, It is manifested by marked thrombocytopenia in patients with a giant hemangioma, without significant alterations in the bone marrow except for an excess of megakaryocytes. The picture is typical of the newborn and normalizes with the removal of the hemangioma.
Kennedy syndrome . Syndrome n and urooftalmoló g i c or or r i g e n tum or ral. It is characterized by unilateral blindness and papilledema on the opposite side. It is caused by tumors of the frontal lobe, or meningiomas of the sphenoid crest, which progressively compress an optic nerve (ipsilateral blindness) and obstruct venous return on the opposite side (contralateral papilledema).
Kimmelstiel-Wilson syndrome . Syndrome of d i to b et e s s c Arina to s or c iado to hypertension or n l and whether or n is r in to the e s d e g e n e rativa s . The latter consist of intercapillary glomerulohyalinosis and are manifested by hematuria, albuminuria, and cylindruria, a nephrotic syndrome, and progressive renal failure.
Kinsbourne syndrome . Syndrome of e pil e psi to m io c lon ic as the infa n cia . It is characterized by myoclonic contractions of the trunk, extremities, and face, with dynamic ataxia and intentional tremor. The cause is unknown, although in 50% of cases there is a hidden neuroblastoma.
Kleine-Levin Syndrome : sind r ome hyper s or m n i to eg river dica. It is characterized by periods of hypersomnolence that last from days to weeks alternating with asymptomatic periods of months to years. During the first, the individual has a voracious appetite and may manifest alterations in mood and sexual behavior (hyperactivity, exhibitionism) along with hallucinations, disorientation and memory disorders. It usually begins in youth and the cause is unknown.
Klinefelter syndrome . Syndrome of c ause GENERI ca c a r a c you ri z to do for endocrine disruption and an or m a l ed s ge and t a l e s.It is distinguished by the eunucoid or weakly masculinized appearance of the patient, gynecomastia, small and hypoplastic testes, hyalinization of the seminiferous tubules with spermatogenesis and preservation of Leydig cells, elevated levels of urinary gonadotrophins (FSH), and decreased androgen secretion. Genetic sex is usually female, there are three or more sets of sex chromosomes (XXY, XXXY or XXYY, XXXXY), and autosomal chromosomes are normal.
Klippel-Feil syndrome . Syndrome produced by the congenital absence of cervical vertebrae. Its basic features are: a) marked shortening or virtual absence of the neck, such that the head seems to rest directly on the upper portion of the trunk; b) inability to turn head; c) extremely low implantation of the nape hair, and d) the radiographs show a decrease in the number of cervical vertebrae, consolidation of the existing vertebrae in a single block, and opening of the posterior vertebral arches (spina bifida).
Klippel-Trenaunay-Weber syndrome . Syndrome associated with unilateral malformations of congenital origin. The picture usually affects one limb and is characterized by exaggerated growth of the limb due to bone and soft tissue hypertrophy, often with syndactyly or other malformations, and by the coexistence of vascular anomalies such as cutaneous hemangiomas, dilations varicose veins or flaming nevi. The cause has not yet been established.
Konig syndrome . Syndrome of a picture of intermittent pain in the right iliac fossa with alterations in intestinal transit. It is characterized by the intermittent appearance of manifestations at the level of the right iliac fossa (colicky pain and localized distension, followed by rumbling that rapidly alleviate the symptoms) and by periods of constipation that alternate with others of diarrhea. It is observed in inflammatory or neoplastic processes that affect the cecum or neighboring areas.
Korsakoff syndrome . Confabulatory amnesic psychosis syndrome. It is observed in chronic alcoholics and is characterized by orientation disorders, susceptibility to external stimuli and suggestion, hallucinations, confabulation, and mainly amnesia, which is disproportionate to the other mental and behavioral manifestations. There are usually polyneuritic alterations (pendulous hand, etc.).
Koshevnicoff syndrome . Syndrome of certain severe forms of motor epilepsy. It includes repeated seizures, hypertemia, delirium, loss of consciousness and paralysis of variable distribution, together with prostration and persistence of myoclonic movements in the intercritical periods.
Kwashiorkor syndrome . Childhood severe caloric protein malnutrition syndrome. It is seen in young children and is characterized by marked anorexia, edema and hypoalbuminaemia, skin changes (depigmentation with erythematous spots, dermatoses, various), discoloration in hair bands, poor appearance, and lack of emotional response. It is due to deficiencies, multiple and prolonged diets.
Crocodile tear syndrome . see Bogorad syndrome .
Landry-Guillain-Barré syndrome . see Guillain-Barre syndrome .
Laubry-Soule syndrome . Digestive syndrome associated with myocardial infarction. It is observed in some patients with myocardial infarction and is manifested by localized meteorism at the level of the left corner of the colon and the gastric chamber. It is considered a reflex phenomenon.
Launois syndrome . Syndrome accompanying certain forms of hyperpituitarism. It is a picture of gigantism that begins before the closure of the epiphyses, usually at puberty, and that results from a hypersecretion of pituitary hormones of tumor origin (eosinophilic adenomas, and sometimes chromophobic adenomas).
Laurence-Moon-Biedl syndrome . Hereditary syndrome produced by hypothalamic dysfunction. Its components are those of Frohlich's adiposogenital syndrome (feminoid obesity, sexual infantilism, mental retardation, etc.), to which retinitis pigmentosa and polydactyly are added. There are no alterations in the adenohypophysis and the condition is of genetic origin.
Milk-alkali syndrome . Hypercalcemia syndrome due to prolonged ingestion of absorbable alkalis. Its manifestations include pruritus, muscle weakness and rheumatiform pain, polydipsia and polyuria, hypercalcemia without hypercalciuria, and generalized calcinosis that can eventually lead to kidney failure. The picture is typical of ulcers who consume large amounts of milk and alkalis, and its incidence has decreased notably since the introduction of non-absorbable antacids.
Leriche syndrome . Syndrome of chronic obstruction of the terminal portion of the abdominal aorta. It is characterized by paleness and cooling of the lower limbs, weakness of the muscles of the hips, legs, or ankles during walking or exercise, sexual impotence, and absence of femoral pulses.
Lermoyez syndrome . Syndrome of certain transitory alterations of the auditory and vestibular function. It includes tinnitus and sudden-onset deafness, which precede a vertigo attack and diminish once vertigo is established. It is attributed to sustained spasms of the internal auditory artery.
Lewis syndrome . Hereditary syndrome due to congenital abnormalities in carbohydrate metabolism. It includes mental retardation of variable degree, neurological disorders (particularly seizures), and hypoglycemic seizures that predominate at night. The cause is a congenital deficiency of hepatic glucogensynthetase.
Lichtheim syndrome . Subacute combined degeneration syndrome of the spinal cord. It is manifested by paresthesias; ataxia, unsteady gait, and later spastic paraplegia. It is seen in idiopathic or Biermer's pernicious anemia and is due to degeneration of the lateral and posterior columns of the spinal cord
Liddle syndrome . Pseudohyperaldoseronism syndrome. It is characterized by hypertension and hypochloraemic alkalosis, with negligible aldosterone secretion. It is hereditary, probably autosomal dominant, and the cause is unknown.
Lightwood syndrome . Syndrome of the inability of the kidney to excrete sufficiently acidic urine. It manifests as a persistent metabolic acidosis with hyperchloremia, and eventually with potassium depletion and hypercalciuria. It is due to a primary defect in the function of the renal tubules (of a familial type or of sporadic appearance) or to a dysfunction secondary to states of dysproteinemia, hypervitaminosis D, hyperthyroidism, etc.
Mucocutaneous lymphogranular syndrome . Confluent dermatitis syndrome of childhood. It begins as an acute febrile picture with polyadenopathy, to which is added a maculoerythematous eruption that is characterized by being confluent and swelling, predominantly in the hands and feet and by the abundant scaling that follows the resolution of the lesions. It was first observed in Japan and its cause is unknown.
Loffler syndrome . Pulmonary syndrome with diffuse allergic manifestations. It is characterized by asthma dyspnea with a severe and rebellious cough, mild or moderate leukocytosis with marked eosinophilia, and the presence of multiple pulmonary granulomas that manifest as diffuse, fleeting infiltrates on radiographs of the chest. The etiology of the condition is not yet known precisely.
Louis-Bar syndrome . Ataxia-telangiectasia syndrome. It is characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, frequent infections of the sinuses and lungs, and abnormal eye movements. There is usually an immune deficiency. It is a hereditary condition that is transmitted as an autosomal recessive trait.
Lown-Ganong-Levine syndrome . Electrocardiographic and clinical syndrome of abnormal atrioventricular excitation. The clinical manifestations are the same as those of Wolff-Parkinson-White syndrome, but unlike this, the only electrocardiographic abnormality is a shortening of the RP.
Lubarsch-Pick syndrome . Congenital syndrome produced by deposit of amyloid substance. It is characterized by the deposition of maternal, atypical amyloid in the skin and muscle tissue, with scleroderma and macroglossia. The cause is unknown.
Lutembacher syndrome . Syndrome associated with congenital cardiac malformations. It designates the combination of atrial septal defect (due to persistence of the foramen ovale), mitral stenosis, and secondary hypertrophy of the left atrium.
Lyell syndrome . Toxic epidermal necrolysis syndrome. It consists of a severe exfoliative dermatitis, preceded by erythema and large blisters. It is usually infectious (staphylococci) in childhood or toxic (due to drugs) in adults.
Alveolar full syndrome . Radiological syndrome of alveolar space occupation. It is characterized by the presence of moderately dense fuzzy nodular images, with diffuse boundaries, which tend to converge in nappa and involve a lung lobe or segment, or which are uniformly distributed in both lung fields. It is observed in bronchopneumonia or in the initial phase of pneumonia, alveolar cancer, acute lung edema, and so on.
Precocious macrogenitosomy syndrome . see Pineal syndrome .
Mallory-Weiss syndrome . Esophageal rupture syndrome due to stress. It is a picture of hematemesis, often massive, that occurs in patients who have been with repeated and persistent vomiting. It occurs from longitudinal lacerations in the vicinity of the esophagogastric junction.
Jaw-wink syndrome . see Gunn syndrome .
Marcus Gunn syndrome. see Gunn syndrome .
Marchiafava-Micheli syndrome . Paroxysmal nocturnal hemoglobinuria syndrome. It is characterized by episodes of paroxysmal hemolysis with hemoglobinuria, usually nocturnal, increased plasma hemoglobin, and sometimes leukopenia and thrombocytopenia. It is prevalent in young adults and is due to a mutation in bone marrow stem cells, with an exaggerated sensitivity to complement.
Marfan syndrome . Syndrome produced by congenital anomalies of the fibrous elements of the connective tissue.Typically it is manifested by: a) tall stature, leanness that is sometimes extreme and thorax of abnormal conformation; b) limbs graceful and disproportionately long in relation to the trunk, which is particularly evident in the more distal segments (aranodactyly); c) scaphocephaly with ogival palate and acetabulum protrusion; d) ocular abnormalities, including ectopia of the lens, corneal opacities, and occasionally myopia and glaucoma; e) muscle weakness, hypermobility of the joints and different bone deformations, and f) a marked tendency to develop aortic insufficiency or aortic aneurysms, as a consequence of the congenital weakness of the middle layer of this vessel. It is a genetic picture that is transmitted as an autosomal dominant trait.
Marie-Bamberger syndrome . Hypertrophic pneumatic osteoarthropathy syndrome. It is secondary to chronic lung disease and is characterized by hyperemia and edema of the periosteum, with neoformation of the bone matrix and subsequent calcification. This produces a symmetrical thickening in the wrists and ankles, metacarpals and metatarsals, and the extremities of the long bones, there are usually “drumstick fingers”. The causal mechanism has not yet been clarified.
Maroteaux-Lamy syndrome . Mucopolysaccharidosis syndrome VI. Its basic characteristics are: a) facies with coarse features, although less accentuated than in Hurler syndrome, with short height, neck and trunk; b) hepatosplenomegaly and a tendency to develop hernias, and c) absence of mental retardation, and slow and late development of corneal opacities. The condition, which also has a mild form, is due to a deficiency of aryl sulfatase B.
Mayer-Rokitansky-Kuster syndrome . Primary amenorrhea syndrome of genetic origin. It is characterized by the absence or marked hypoplasia of the vagina, a rudimentary uterus sometimes associated with abnormalities of the urinary system and occasionally with skeletal abnormalities. Both secondary sexual characteristics and ovarian function are normal, and if the rudimentary uterus has an endometrium, there may even be menstrual bleeding that, when retained, produces abdominal pain of cyclical appearance.
Meigs syndrome . Syndrome associated with the presence of ovarian tumors. It is the combination of ascites and hydrothorax with an ovarian tumor, usually a fibroma.
Melkersson-Rosenthal syndrome . Syndrome of facial paralysis with chronic swelling of the soft tissues. It is a recurrent facial paralysis associated with chronic, persistent facial edema, which predominates on the lips and tongue. The cause has not been clarified.
Ménétrier syndrome . Thoracic duct obstruction syndrome. Hard edema, affecting the lower limbs, the lower abdomen and the left hemithorax and arm, associated with hydrothorax or chylothorax and peritoneal effusion. The cause is usually tumor.
Méniere syndrome . Recurrent vertigo syndrome, accompanied by tinnitus and deafness. It usually begins around 50 years of age, and is manifested by acute attacks of vertigo, deafness that is usually unilateral, and tinnitus in the affected ear. The classic form is due to a labyrinthine hemorrhage, while in other cases it results from degenerative processes in the inner ear.
Meningeal syndrome . Neurological syndrome resulting from irritation of the meningeal membranes. The picture is typical and is manifested by: a) photophobia and intense, persistent and rebellious headache to any analgesic medication; b) easy, central gushing vomiting and persistent constipation; c) stiffness in extension of the trunk and neck, with cutaneous hyperesthesia and muscular hyperreflexia; d) disorders of consciousness, seizures, coma, e) quantitative and qualitative alterations of the cerebrospinal fluid, (hypertension, pleocytosis, presence of blood or purulent material, etc.). It is produced by hemorrhages, infections and irritative processes.
Myasthenia gravis syndrome . Muscle weakness syndrome related to alterations of the neuromuscular junction that prevent the normal transmission of nerve impulses.Its basic features are: a) onset, usually subacute or insidious; b) muscular weakness that predominates in the eye muscles (without affecting the pupils), in the muscles, facial, laryngeal and pharyngeal, and in the diaphragm; c) quick and easy muscle fatigue when repeating movements or trying to maintain a posture, partial recovery after a period of rest and noticeable improvement in response to the administration of agents, cholinergics (neostigmine), and d) there is no muscle atrophy or alteration of reflexes, or other neurological abnormalities, and the main risk is the ease with which the individual develops episodes; respiratory failure. There is an idiopathic form, the most frequent, and others that are associated with thymus tumors, hyperthyroid conditions, and collagen diseases.
Myeloproliferative syndrome . General name for a group of conditions characterized by the uncontrolled proliferation of cell types in the bone marrow. It defines the signs and symptoms of non-acute evolutionary processes that are manifested by the predominant proliferation, rather than isolated, of cells of the granulocytic, megakaryocytic, erythroid line, and eventually, of fibroblasts. They are the following: a) chronic granulocytic leukemia, which presents with great splenomegaly and circulating granulocyte numbers that often reach 200,000 per mm 3; b) essential hemorrhagic thrombocythemia, where megakaryocytes clearly predominate and splenomegaly is moderate; c) polycythemia vera, characterized by an increase in the number of circulating cells of the three series, but mainly at the expense of erythrocytes and with hematocrits that generally exceed 60%, and d) agnogenic myeloid metaplasia or myeloid metaplasia -myelofibrosis, a condition in which granulocytes and megakaryocytes predominate but with a simultaneous progressive fibrosis of the bone marrow, so that in the advanced stages when this fibrosis has been completed, cell proliferation takes place in extramedullary foci (metaplasia) and explains the remarkable hypertrophy of the liver and spleen that these patients present. These four paintings have in common,
Milroy-Nonne-Melge syndrome . Bilateral chronic lymphedema syndrome of the lower limbs. It is characterized by chronic and progressive edema of the legs, which begins in the first stages of life, sometimes associated with hemangiomas and lymphangiomas. The condition is hereditary, autosomal dominant; it predominates in women and is due to congenital lymphatic hypoplasia.
Mikulicz syndrome . Syndrome of hypertrophy of the salivary and lacrimal glands. Symmetrical and usually insidious and painless enlargement of the salivary and lacrimal glands, which is accompanied by a concomitant partial or complete suppression of the secretion of tears and saliva. There are idiopathic forms and others that appear in patients with sarcoidosis, lymphomas, specific infections, etc. The etiology is unknown.
Milkman syndrome . Syndrome typical of certain forms of severe osteoporosis of mature age. It is observed predominantly in women and is characterized by a) diffuse decalcification with significant skeletal alterations (marked kyphosis, vertebral crushing, bone deformation); b) back pain and pain in the lower limbs, often of the radicular type; c) increasing weakness and asthenia, and d) characteristic abnormalities on bone radiographs (presence of transverse radiolucent bands surrounded by an opaque border). The etiology of the condition is unknown.
Minkowski-Chauffard syndrome . Hereditary spherocytosis syndrome. It is characterized by mild to moderate anemia, splenomegaly, and jaundice due to increased unconjugated bilirubin, due to the presence of spherocytes and increased red blood cell fragility. It is transmitted as an autosomal dominant trait.
Möbius syndrome . Neurological syndrome of dysgenesis of the nuclei of the cranial nerves. It is manifested by types and degrees of paralysis or paresis that vary according to the greater or lesser involvement of the different cranial nerves, expressionlessness of the face, ocular motility alterations, difficulty in sucking, dysphagia; dysarthria, etc. Occasionally these patients present with syndactyly and muscle group atrophy. The cause is unknown.
Von Monakow syndrome . Neurological syndrome of anterior choroidal artery occlusion. Alternate hemiplegia accompanied by hemianopia and homonymous hemianesthesia or hemihypoesthesia due to lesions affecting the posterior arm of the internal capsule.
Mondor syndrome . Syndrome of the severe forms of septic abortion. It is characterized by fever spikes, rapidly increasing jaundice, marked hypotensive hemoglobinuria, and oliguria that almost always progresses to acute renal failure. The facies is usually typical: conjunctival jaundice pale cheeks and perioral cyanosis.
Morgani-Stewart-Morel syndrome . Internal frontal hyperostosis syndrome. It consists of thickening of the internal table of the frontal bone that affects almost exclusively the female sex, close to menopause, and is accompanied by obesity, hypertrichosis and different neuropsychic manifestations. It is of unknown cause.
Morquio syndrome , syndrome mucopolysaccharidosis IV. It is distinguished by: a) facies with moderately coarse features, broad nose; wide mouth and dental alterations; b) short height and neck, marked kyphosis, and anteroposterior widening of the thorax; c) genu valgum with duck gait; d) hepatosplenomegaly and eventually aortic insufficiency, and e) presence of corneal opacities, but without mental retardation. It is transmitted as an autosomal recessive trait and is due to a deficiency of chondroitin sulfate sulfatase.
Moschcowitz syndrome . Purpuric syndrome with disseminated thrombosis. The picture is that of a severe hemolytic anemia of the extracorpuscular type, associated with thrombocytopenic purpura and multiple neurological disorders (cranial nerve palsy, cerebellar signs, vertigo and seizures). The cause is unknown.
Mucopolysaccharidosis syndrome . Congenital deficiency syndrome of specific enzymes of mucopolysaccharidosis metabolism.It includes a set of clinical pictures that have in common: a) a congenital deficiency of some of the essential enzymes for the degradation of three types of mucopolysaccharides: heparan sulfate, dermatan sulfate and keratan sulfate; b) the progressive nature of the condition, whose manifestations are due to the increasing accumulation of mucopolysaccharides in the different body systems, and c) the existence of certain basic similarities regarding the phenotype: gross features, dysostosis, multiple, rigidity joint, hepatosplenomegaly, excretion of mucopolysaccharides in urine, etc. Instead, they differ from each other by the severity of the skeletal alterations, by the presence or absence of certain particular components (mental retardation, opacities in the cornea), by the seriousness of the prognosis, and due to the enzymatic deficiency of each case. From combinations of these elements, the general group of mucopolysaccharidoses has been subdivided into the following particular types: type IH, or Hurler syndrome; type IS, or Scheie syndrome (formerly: type V); type II or Hunter syndrome; type III, or Sanfilippo syndrome; type IV or Morquio syndrome; type VI, or Maroteaux-Lamy syndrome; type VII, or Sly syndrome; and type VIII or Di Ferrante syndrome. or Sanfilippo syndrome; type IV or Morquio syndrome; type VI, or Maroteaux-Lamy syndrome; type VII, or Sly syndrome; and type VIII or Di Ferrante syndrome. or Sanfilippo syndrome; type IV or Morquio syndrome; type VI, or Maroteaux-Lamy syndrome; type VII, or Sly syndrome; and type VIII or Di Ferrante syndrome.
Sudden Infant Death Syndrome . Name by which cases of unexpected and unexplained death of an infant are designated. Typically, this is the case of the child who falls asleep peacefully at night, and the next morning he is found dead in his crib (cribcot-death, from the Saxon authors), without any explanation or findings to justify it, in autopsy studies. The cause is unknown, although different mechanisms have been postulated that could be responsible for a condition of this type: the child's inability, due to immaturity, to increase ventilation in response to moderate respiratory obstructions; intervention of a condition such as adult sleep apnea; triggering of cardiac arrhythmias and others.
Munchhausen syndrome . Fabulation syndrome in a histrionic personality. It is characteristic of individuals who request to be treated in hospitals claiming to be suffering from an acute illness, about which they offer a coherent, often dramatic, but false story, and who reiterate this behavior in different situations in their lives.
Cystic stump syndrome . Postoperative syndrome of cholecystectomies. It includes a set of more or less imprecise symptoms, similar to those of post-cholecystectomy syndrome except for the absence of jaundice, which are associated with a dilation of the residual stump and are attributed to reflex phenomena originating in this area.
Naegeli syndrome . Syndrome characterized by the association of hyperkeratosis, pigment abnormalities and other skin disorders. It includes palmoplantar hyperkeratosis, reticulated pigmentation of the skin, anhidrosis or hypohidrosis, and sometimes dental changes. It is hereditary and is transmitted as an autosomal dominant trait.
Nelson syndrome . Syndrome following certain treatments for Cushing's disease. It is seen in patients with Cushing's disease who underwent bilateral adrenalectomy with irradiation of the pituitary. It consists of the development of a more aggressive ACTH-secreting tumor that produces serious alterations of the visual field due to local invasion.
Multiple endocrine neoplasms syndrome . Set of manifestations that result from the simultaneous hyperfunction of several different endocrine systems. It designates a variety of clinical pictures associated with the presence of hyperplasias, or multiple endocrine tumors (pancreatic; parathyroid, pituitary, thyroid and adrenal, as well as neuromas) and whose manifestations are the result of hyperfunction of the corresponding glands. Although the varieties and possible theoretical combinations are many, the specific forms of presentation have made it possible to establish three different groups within the syndrome of multiple endocrine neoplasms: type I or Wermer syndrome.in which thyroid, adrenal and pituitary tumors, parathyroid hyperplasia and non-insulin rumors of the islets of Langerhans occur, and to which the Zollinger-Ellison syndrome, previously considered as a separate entity, tends to be incorporated; the type II or Sipple syndrome, characterized by the association of medullary thyroid carcinoma; with pheochromocytomas and parathyroid hyperplasia; and type III or multiple mucosal neuroma syndrome, a combination of a pheochromocytoma and a medullary carcinoma of the thyroid with numerous mucosal neuromas and different body abnormalities.
Neuroanemic syndrome . see Lichtheim syndrome .
Multiple mucous neuroma syndrome . Type III syndrome of multiple endocrine neoplasms. It is defined by the association of thyroid and adrenal tumors (medullary thyroid carcinoma, pheochromocytoma), by the development of multiple neuromas in the mucosa (mouth, conjunctiva, esophagus, digestive tract) and by the presence of body malformations (habit similar to of Marfan syndrome, kyphoscoliosis, genu valgum, pes cavus, prognathism of soft tissues, etc.).
Retrobulbar neuropathy syndrome . Reversible blindness syndrome of unknown etiology It manifests itself in adolescents or young adults by a sudden decrease in visual acuity in one eye, which rapidly evolves to blindness, followed by the same picture a few weeks later in the other eye. The pupil and retina are normal and there are no neurological alterations to justify the disorder. Most patients recover spontaneously, with no sequelae or with minor or pinpoint defects in the visual field.
Basal cell nevus syndrome . see Gorlin-Goltz syndrome .
Sick sinus node syndrome . see Sinus node disease syndrome .
 Noonan syndrome |
Noonan syndrome . Hereditary syndrome characterized by hypogonadism, cardiac malformations and other bodily abnormalities. Its basic features are: a) height and short neck, with hypertelorism, low implantation of the hair and a shield thorax; b) hypogonadism and cryptorchidism; c) congenital pulmonary stenosis and occasionally, aortic stenosis, and d) in some cases, mild or moderate mental retardation. There are no alterations in the karyotype.
Nothnagel syndrome . Neurological brain stem syndrome with cerebellar disorders and third cranial nerve involvement. It is characterized by partial unilateral or bilateral paralysis of ocular motility (III cranial nerve), associated with cerebellar ataxia (lesion of the superior cerebellar peduncle). It is usually caused by tumors of the roof of the midbrain.
Syndrome of the ambiguous nucleus and the spinothalamic tract . see Avellis syndrome .
Deiters nucleus syndrome . see Bonnier syndrome .
Acute arterial obstruction syndrome . Syndrome of the sudden interruption of the arterial flow to a sector of the organism. It is characterized by pain, coldness, paleness, anesthesia, functional impotence and disappearance of the peripheral pulse. It is due to a spasm or the presence of an organic obstacle in an artery (thrombosis, embolism, exogenous compression).
Migraine ophthalmoplegia syndrome . Transient ophthalmological syndrome associated with migraine attacks. It occurs in young adults in the form of one or more unilateral attacks of paralysis, of eye movements (III and VI cranial nerves) associated with a migraine, a condition that is not different at all from the classic form of migraine attack. Patients heal spontaneously and the cause is unknown, although the syndrome has been attributed to prolonged spasm of some branch of the ophthalmic artery.
Ogilvie syndrome . Syndrome of apparent obstruction of the colon. It is manifested by states of persistent contracture of the muscles of the large intestine, with no organic cause to justify them. It is attributed to abnormalities of sympathetic innervation.
Oppenheim syndrome . Syndrome of the conditions that evolve with muscular flaccidity in the pediatric patient. General name, purely clinical and independent of etiology, of all conditions of the newborn and childhood that present with decreased tone of skeletal muscles, and of which Werdnig-Hoffinann syndrome is the best known entity.
Ostrum-Frost syndrome . Syndrome characterized by craniocervical and shoulder girdle malformations . It is a triad consisting of congenital synostosis of the neck, occipital intussusception with protrusion of the cervical vertebrae in the posterior fossa (platybasia) and congenital elevation of the scapula (Sprengel's deformity).
Polycystic ovarian syndrome . see Stein-Leventhal syndrome
Pancoast syndrome . Neuromuscular syndrome of vertex lung tumors. It is defined as a picture of rheumatic pain in the upper limb, sometimes with focal muscular atrophy, accompanied by miosis and paralysis of the levator upper eyelid. It is typical of neoplasms close to the pulmonary apex that invade the roots of the brachial plexus.
Chronic pancreatitis syndrome . Syndrome of chronic inflammation of the pancreas. It is manifested by: a) recurrent episodes of mild to moderately intense abdominal pain of variable duration (hours or days); b) steatorrhea and latent or manifest diabetes, of insidious installation and progressive evolution, and c) presence of calcium deposits in the entire organ, visible on radiographs. The condition predominates in alcoholic, obese or bile duct lithiasis individuals, although there are also idiopathic forms.
Hereditary pancreatitis syndrome . Infant pancreatitis syndrome. It is defined by the appearance of repeated attacks of intense abdominal pain, lasting from days to weeks, with increased plasma levels of amylase and lipase, and by the progressive development of the chronic manifestations of the disease: diabetes, steatorrhea and calcification of the pancreas. The condition is hereditary and is transmitted as an autosomal dominant trait with incomplete penetration.
Papp-Parkinson syndrome . Repetitive paroxysmal tachycardia syndrome. It is characterized by the appearance of very numerous short episodes of paroxysmal atrial tachyarrhythmia (supraventricular tachycardia, atrial flutter, atrial fibrillation), separated by also short intervals of sinus rhythm with frequent atrial extrasystoles. The condition is of unknown cause, can be repeated up to 1000 times per day, and leads the individual to a state of virtual disability.
Familial periodic paralysis syndrome . Syndrome of intermittent paralysis with associated hypokalemia. It is characterized by recurrent attacks of flaccid paralysis of the limb muscles, with tendon areflexia and lack of muscle response to electrical stimulation. The attacks usually occur at night, often after eating meals rich in carbohydrates, and are typically accompanied by a marked decrease in plasma potassium levels. It is a family condition, with a mechanism that is not well known, which allows the individual to develop a normal life in the intercritical periods.
Foix paramedian syndrome . see Dejerine Syndrome , def. 2.
Paraneoplastic syndrome . Set of manifestations associated with the presence of a malignant tumor in organs and systems not directly affected by neoplastic growth.It includes different conditions that often precede or accompany the clinical manifestation of the tumor, and include: a) endocrine syndromes (Cushing's, gynecomastia, hyperthyroidism, hypoglycemia, hyperpigmentation, hyperparathyroidism, etc.) b) rheumatiform (polymyositis, hypertrophic osteoarthropathy, polyarthritis, etc.); c) neurological (cerebellar or cerebral disorders), d) hematological (thrombosis, disseminated) and others. Examples of this are the association of hyperparathyroidism with kidney, pancreatic or ovarian tumors; gynecomastia or hyperpigmentation with lung carcinomas; hyperthyroidism with choriocarcinoma or with embryonal carcinoma of the testicle; and acanthosis nigricans with lymphomas or adenocarcinomas.
Raeder's paratrigeminal syndrome . Unilateral pain syndrome of the face. It is characterized by unilateral crises of intense facial pain, located in the maxillary and frontotemporal region, and accompanied by hypoesthesia and pupillary inequality. Unlike trigeminal neuralgia, it is caused by organic lesions (tumors; granulomas).
Parinaud syndrome , syndrome neurological, of the brainstem with cranial nerves commitment. It is characterized by upward gaze palsy, fixed pupils, and diverging eyes. It is caused by injury to the anterior quadrigeminal tubercles and the supranuclear upward gaze coordination mechanism, and the cause is usually a pinealoma.
Parkinsonian syndrome . S índrome of neurological paralysis agitans.It is an insidious onset and progressive evolution whose basic features are: a) typical expressionless, seborrheic facies with exaggerated salivation; b) generalized muscle stiffness; c) involuntary thick tremor, present at rest but which disappears during sleep and tends to disappear with movements; d) also characteristic gait (festive gait), fast, with short steps and with the body leaning forward. There are no alterations in the intellect or the sense organs. It is caused by injury to the pigmented nuclei of the brain stem (substantia nigra, locus coeruleus) and is observed as a pure entity in Parkinson's disease, and associated with other manifestations in conditions of different etiology (atherosclerotic parkinsonism, post-encephalitic parkinsonism, etc. ).
Paterson-Kelly syndrome . see Plummer-Vinson syndrome .
Claude's pedicle syndrome . Neurological syndrome of the brainstem with involvement of the third cranial nerve and cerebellar alterations and of the articulation of the word. It is characterized by paralysis of ocular motility on the side of the lesion (III cranial nerve), accompanied by hemiasynergia on the opposite side (lesion of the red nucleus) and dysarthria. It is caused by aneurysms, thrombosis, or tumors of the brain roof.
Pellegrini-Stieda syndrome . Knee joint dysfunction syndrome. Chronic pain, swelling and limitation of knee movements, with calcification of the internal collateral ligament of said joint. It is a distant sequel to trauma or traumatic bruising.
Pende syndrome . Global hyperfunction syndrome of the adenohypophysis. It is manifested by: a) moderate increase in height; b) tendency to obesity, preferably at the level of the hips and thighs; c) mammary hyperplasia in women and gynecomastia in men; d) signs of metabolic craniopathy, and e) skin striae: similar to those of Cushing's disease. There is no high blood pressure and the cause of the disorder is unknown.
Pendred syndrome . Hereditary syndrome characterized by deafness and alterations in thyroid function. It is manifested by congenital bilateral deafness and by the later development in early childhood of a non-hypothyroid goiter. The disorder is seen in regions where there is no endemic goiter and its cause is unknown, although it is attributed to a congenital inability to synthesize thyroid hormones normally.
Penfleld syndrome . Episodic neurovegetative hyperfunction syndrome associated with cerebral dysrhythmia. It is characterized by brief fits of restlessness, with vasomotor phenomena in the sympathetic cervical territory, arterial hypertension, diaphoresis lacrimation, pupillary myiosis or mydriasis, tachycardia, bradypnea, and sometimes loss of consciousness. It was attributed by Penfield to epileptic discharges originating in the dorsal nucleus of the thalamus.
Posterior perisylvian syndrome . see Sensory Aphasia Syndrome
Peutz-Jeghers syndrome . Hereditary syndrome characterized by skin changes and a tendency to develop malignant processes. It is manifested by the presence of pigmented macules on the lips, the buccal mucosa, and the fingers, and by an intestinal polyposis that predominates in the small intestine. The condition is transmitted as an autosomal dominant trait and an increased tendency for the development of gastric, duodenal, and colon adenocarcinomas has been shown.
Pick's syndrome . Chronic constrictive pericarditis syndrome. It is a picture of chronic intrapericardial hypertension, consecutive to rheumatic or tuberculous pericarditis, in which there is liver fibrosis and jugular engorgement, but without heartbeat or respiratory oscillations. Fluoroscopy does not show the characteristic movements of cardiac activity ("still heart").
 Pickwick syndrome |
Pickwick syndrome . Pulmonary hypoventilation syndrome associated with obesity. It is basically characterized by obesity, marked daytime sleepiness, polycythemia, and exaggerated appetite, with increased arterial PC0 2 and no signs of underlying organic lung disease; in addition, there is usually cyanosis and periodic nocturnal respiration, and eventually, signs of right heart failure. The manifestations improve markedly, when the patient reduces his weight.
Restless legs syndrome . Syndrome of one of the varieties of insomnia.It is characterized by: a) the virtually irrepressible need of the individual to move their legs when immobile or sitting, and very particularly when lying down and trying to sleep; b) the explanation he provides about these movements, which he relates to a deep, intense and extremely unpleasant discomfort he experiences in his ankles, and from which he tries to free himself by moving his legs; c) the disappearance of the sensation after having slept, and d) in all cases, the possibility of recording, during sleep, stereotyped rhythmic movements, dorsiflexion of the feet and toes (periodic sleep movements) , which are characteristic of different types of insomnia. The cause is unknown, although it is attributed to actual neurophysiological alterations, although still incompletely known.
Pineal syndrome . Syndrome in which certain tumors of the thymus appear. It is characterized by early abnormal growth of long bones, early development of the external genitalia and sexual function, and intracranial hypertension.
Orbital floor syndrome. see Dejean syndrome .
Plummer-Vinson syndrome . Idiopathic hypochromic anemia syndrome with achilia. It is observed almost exclusively in women and is manifested by: a) hypochromic anemia that responds satisfactorily to the administration of high amounts of iron; b) gastric achilia; c) atrophy of the mucosa of the mouth, pharynx and upper esophagus, with dysphagia and glossitis; and characteristically, d) presence of a crescent-shaped fold in the cricopharyngeal region. The etiology of the condition is unknown.
Postcholecystectomy syndrome . Vaguely defined syndrome in certain patients undergoing cholecystectomy.A set of symptoms that are often vague and ill-defined, including diffuse or colicky pain or discomfort in the right upper quadrant of the abdomen, food intolerances, a feeling of fullness, etc., and occasionally jaundice. In a strict sense, the name should be applied only to those cases in which it is shown that the manifestations are the consequence of incomplete operations (residual stones), of intraoperative omissions (neoplasms that went unnoticed) or of specific complications (postoperative stenosis). However, by custom rather than by extension, it is also applied to patients with complaints that suggest an emotional or functional background just because they have undergone surgery, and to others who complain of the same symptoms they had before the surgery. intervention,
Postcommissurotomy syndrome . Postoperative syndrome far from commissurotomy due to mitral stenosis. It is a relatively frequent picture in this type of operation, which appears one to three months after the intervention and is manifested by fever, retrosternal pain, diffuse, often migratory arthralgias, cardiac arrhythmias; signs of polyserositis (pericarditis, pleurisy), etc. The cause has not been precisely established.
Postphlebitic syndrome . see Post-thrombotic syndrome .
Postgastrectomy syndrome . see Rapid emptying syndrome
Post myocardial infarction syndrome . see Dressler's syndrome .
Post-radiation syndrome . see Radiation syndrome .
Post-perfusion syndrome . Cytomegalovirus infection syndrome. It is an acute febrile picture with hepatomegaly, splenomegaly and the presence of atypical lymphocytes, which was initially observed in patients undergoing cardiac interventions with extracorporeal circulation. Later it was shown that the causative agent was a cytomegalovirus and that the possible routes of transmission were many.
Postachycardial syndrome . Residual electrocardiographic syndrome of ventricular tachycardias. It is observed after prolonged attacks and is characterized by the appearance of widened and inverted T waves that persist for a variable time (hours or days) before finally disappearing. It is a manifestation of the functional recovery process and lacks, by itself, pathological significance.
Post-thrombotic syndrome . Chronic syndrome of venous thrombosis of the lower limbs. It is a dermatological condition that is observed in patients who have suffered repeated thrombosis, and is characterized by a first phase in which edema and subcutaneous hemorrhages predominate; a second phase with hemosiderin hyperpigmentation, subcutaneous fibrosis, skin atrophy and lymphatic obstruction; and a third phase in which ulcers appear: chronic, recurrent and difficult to heal.
Pourfour du Petit syndrome. Sympathetic cervical irritation syndrome. It is the opposite of Horner's syndrome and is manifested by exophthalmia, mydriasis, and increased palpebral fissure. It is the result of an irritative phenomenon, usually due to compression or tumor infiltration.
Premotor syndrome . Neurological syndrome of premotor cerebral cortex lesions. It manifests as spastic hemiplegia with osteotendinous hyperreflexia, loss or diminution of the ability to perform fine movements, transient vasomotor disorders, and attitudes of the compulsive apprehension type. It is produced, almost always, by expansive tumor processes.
Profichet syndrome . Syndrome characterized by subcutaneous calcifications and muscular alterations. It is manifested by the gradual formation of calcium nodules under the skin, especially around the large joints, followed by muscle atrophy, skin ulcerations, and neurological disorders. It predominates in young individuals and the cause has not been established.
Superior pontine syndrome . see Raymond-Cestan syndrome .
Putnam-Dana syndrome . see Lichtheim syndrome .
QT syndrome. Syndrome characterized by electrocardiographic abnormalities and episodes of paroxysmal tachyarrhythmia. It applies to those cases in which an abnormally prolonged QT interval, congenital in nature and present in normal sinus rhythm, is associated with paroxysmal episodes of polymorphic ventricular tachycardia ("torsade de points"), in which the transit from sinus rhythm towards arrhythmia is characterized by the increasingly marked deformation of the successive QRS complexes, until the characteristic pattern of ventricular tachycardia is established. It includes the Jervell and Lange-Nielsen syndromes, of autosomal recessive transmission, accompanied by nervous deafness, and that of Romano-Ward, of autosomal dominant inheritance and without nervous deafness.
Chiasmatic syndrome . Optic chiasm lesions syndrome. It is manifested by headache, dizziness and lightheadedness, progressive loss of vision, often not systematized and depending on the greater or lesser extent to which the different bundles are affected; and visual field reduction. The cause is tumor.
Ramsay-Hunt syndrome . Syndrome of virotic affections of the geniculate ganglion. It is manifested by a severe, unilateral facial paralysis, accompanied by earache and a vesicular eruption, in a bunch, affecting the external auditory canal and the pinna. It is due to the lesion of the geniculate ganglion caused by the herpes zoster virus.
Raymond-Cestan syndrome . Pons injury neurological syndrome. It is characterized, on the opposite side of the lesion, by hemiparesis, hemianesthesia, static tremor, and cerebellar-type asynergy, and by paralysis of the lateralization movements of the gaze that predominates on the side of the lesion. It is usually vascular in origin and is caused by injuries that affect the upper portion of the annular pons.
Raynaud's syndrome . Syndrome characterized by bouts of paroxysmal vasoconstriction.It designates a picture of paroxysmal ischemic episodes, mainly of the fingers of the hands, which characteristically present as a triphasic reaction: first paleness, then cyanosis, and finally redness, often with pain and paresthesia during the first phase. From a clinical point of view and for the same syndrome, it is necessary to separate Raynaud's disease, a benign and symmetric functional disorder that predominates in young women and is usually triggered by cold or emotion, from the secondary Raynaud's phenomenon, which accompanies other pathological processes. The latter has no distinction of sex and age, it can be asymmetric, sometimes it can cause digital ulcers, and it is observed in certain collagenopathies; (scleroderma), in lead and arsenic poisoning, during the administration of different drugs (ergotamine methysergide, propranolol), in hematological diseases; (by cold agglutinins); in occult carcinomas, etc.
Reichmann syndrome . Gastric hypersecretion syndrome. It designates the state characterized by hypersecretion of acid gastric juice, whatever its cause and regardless of eventual complications.
Reifenstein syndrome . Male pseudohermaphroditism syndrome associated with primary hypogonadism. It is characterized by eunucoidism, gynecomastia, and hypospadias, and by atrophy of the seminiferous tubules with the absence of sperm. The disorder is due to abnormalities in the synthesis or peripheral action of testosterone, manifests in post-pubertal age, and is transmitted as an X-linked recessive trait.
Reye syndrome . Childhood paravirotic syndrome with hepatic neurological and renal disorders.It occurs in previously healthy children with upper respiratory tract infections, and is characterized by: a) signs of severe hepatocellular damage, with hypoglycemia, acidosis, hyperammonemia, and a marked increase in serum transaminases; b) severe encephalopathy with vomiting, cerebral edema and rapidly progressive changes in the central nervous system; c) presence of extreme vacuolization of liver cells and renal tubules, and d) high mortality, but with complex recovery in patients who survive. It is observed as a complication of viral infections (due to viruses; influenza A, chickenpox, the Coxsackie group, and others) whose initial course does not differ from the usual for each type of disease, and almost always in patients who have been treated with aspirin. For this reason,
Riley-Day syndrome . Family dysautonomia syndrome. It is characterized by autonomic instability (abnormal perspiration, loss of vasomotor control, labile hypertension), taste disorders, decreased pain and temperature sensations, hyporef1exia, episodic fever, seizures, vomiting, and lack of lacrimation. Most of those affected do not reach adulthood. It would be due to an enzyme deficiency not yet specified.
Subclavian steal syndrome . Ischemia syndrome in the vertebrobasilar territory, due to stenosis -before the birth of the vertebral- of one of the subclavian arteries. It is characterized by the appearance of dizziness, syncope or other manifestations of vertebrobasilar insufficiency, when the patient exercises with the arm on the affected side. In these circumstances, in which the subclavian does not manage to provide the blood that the active limb needs, a compensatory flow inversion occurs in the vertebral system (from the brain it passes to the subclavian, beyond the occlusion, and from there arm) and symptoms appear; that is, the subclavian "steals" the blood that should go to the brain.
Rochon-Duvigneau syndrome . Neurological syndrome of unilateral injury to cranial nerves III, IV, V and VI. It is usually an insidious and progressive condition that leads to complete ophthalmoplegia (cranial nerves III, IV and VI), with anesthesia of the cornea (ophthalmic branch of the V cranial nerve). It is caused by aneurysms, meningiomas, and other tumors that grow in the vicinity of the sphenoid cleft
Roger syndrome . Syndrome of malignant tumors of the esophagus. It designates the picture of continuous salivation that accompanies neoplasms of the esophagus or the esophagopharyngeal region.
Romano-Ward syndrome . see QT Syndrome
Rothmann-Makai syndrome . Syndrome of circumscribed inflammation of the subcutaneous fat tissue. It designates a benign picture of circumscribed panniculitis, manifested by necrosis of fat cells and the formation of adipose granulomas that transform into cystic structures. The cause is unknown.
Rothmund-Thomson syndrome . Familial sclerodermic syndrome with cutaneous telangiectasias and multiple organic alterations. It is characterized by the development of: a) symmetrical, progressive banded sclerosis that predominates on the face and fingers and rapidly leads to stiffness; b) telangiectasias, cutaneous with associated pigmentary alterations; c) cataracts and generalized signs of premature aging, d) trophic ulcerations and e) hypogonadism and other abnormalities. It is transmitted as an autosomal recessive trait.
Syndrome of the imminent rupture of the uterus . see Bandl-Frommel syndrome
Roussy-Lévy syndrome . Neuromuscular syndrome with central and peripheral manifestations. It designates the combination of a progressive muscular atrophy, of neuropathic origin and that predominates in the peroneal region, with cerebellar ataxia and scoliosis of the dorsal and lumbar spine.
Sanfilippo syndrome . Mucopolysaccharidosis syndrome III. It is characterized, within the group of: mucopolysaccharidosis by the notable predominance of neurological alterations compared to somatic ones, which are relatively mild. Its basic features are: a) coarse-featured facies, shaggy eyebrows, a half-open mouth and multiple dental anomalies; b) moderate joint stiffness with minimal skeletal alterations (neck and short fingers); c) hepatosplenomegaly, and d) deafness, severe mental retardation and absence of corneal opacities. It is transmitted as an autosomal dominant trait, due to deficiency of different; enzymes (heparansulfatase, N-acetyl-D-glucosaminidase, etc.) and four different variants have been described: (types A, B, C and D).
Sayre-Kearns syndrome . Syndrome characterized by the association of ocular myopathy, myocardial myopathy and retinitis pigmentosa. It is defined by the development of progressive bilateral ophthalmoplegia that respects the pupil and the muscles of accommodation, accompanied by retinitis pigmentosa and concomitant cardiac myopathy, and by the presence of a striking number of mitochondria in the affected tissues. The cause is unknown.
Scheie syndrome . Mucopolysaccharidosis syndrome IS. It is characterized by: a) coarse facial features, with a large mouth, wide nostrils and shaggy eyebrows; b) joint stiffness that appears around the age of 10, with minor skeletal disorders and aortic insufficiency in later stages of life; c) presence of corneal opacities, and d) normal intelligence. It is transmitted as an autosomal recessive trait and is due, like Hurler syndrome, to a deficiency of alpha-L-iduronidase, but unlike this, affected individuals can have a long survival.
Schmidt syndrome . Neurological syndrome of unilateral lesion of the nuclei ambiguus and accessory of the X cranial nerve. It is defined by the presence of paralysis of the vocal cord, soft palate, and trapezius and sternocleidomastoid muscles, on the same side of the injury. The cause is usually vascular or tumor
Schwachmann syndrome . Syndrome of congenital hypoplasia of the pancreas with hematological abnormalities. The dominant manifestation is a picture of diarrhea with steatorrhea, similar to that observed in cystic fibrosis of the pancreas, accompanied by hematological alterations whose cause lies in the bone marrow (neutropenia, and less frequently, anemia and thrombocytopenia), and overshadowed by breakthrough infections that are often severe. Unlike cystic fibrosis of the pancreas, the concentration of solutes in sweat is normal and there are no respiratory disorders. It is transmitted as an autosomal dominant trait.
Seabright-Bantam syndrome . Pseudohypoparathyroidism syndrome. Its basic features are: a) dwarfism, obesity and mental retardation of variable degree; b) round facies, and short neck, trunk, limbs and fingers; c) presence of disseminated calcifications; d) hypocalcemia, tetany crisis and seizures; and e) parathyroid hyperplasia with elevated levels of circulating parathyroid hormone. The picture is familial of unknown cause, and is due to a primary resistance of the effector organs (bones and kidneys) to the action of endogenous parathyroid hormone.
Selye syndrome . see General adjustment syndrome .
Senear-Usher syndrome . Erythematous pemphigus syndrome. It consists of chronic lesions with a seborrheic appearance, which predominate on the face and trunk and usually do not affect the mucous membranes. The evolution is longer than that of pemphigus vulgaris.
Carotid sinus syndrome . Carotid sinus stimulation syndrome. It is a sudden picture of vertigo, lipothymia or syncope, sometimes with seizures, triggered by accidental mechanical stimulation of a carotid sinus (digital compression, abrupt and exaggerated rotation of the neck). Hypotension is associated with bradycardia, characteristic of vasovagal episodes.
Cavernous sinus syndrome . see Foix syndrome .
Pseudobulbar syndrome . Neurological syndrome associated with diffuse lesions of the central nervous system . It is defined by the presence of multiple neurological and psychic disorders, and in particular of mimicry, swallowing and phonation, produced by lesion of the nuclei of the base, the facial regions of the cerebral cortex or the geniculate bundles. In general, it does not present as a pure syndrome, but as part of a picture of global brain deterioration, produced by diffuse vascular lesions, or by multiple softening foci.
Intermittent pseudoclaudication syndrome . Neurological cause of gait claudication syndrome.It is a picture that is easily confused with that of intermittent claudication of vascular origin (pain that appears during walking and is relieved with rest), but from which it differs due to: a) the relationship between the distance traveled and the appearance of pain , which is not constant as in the case of ischemic claudication, but highly variable, and between the beginning of rest and the disappearance of pain that does not occur quickly, but slowly and in a variable time; b) the distribution of pain, which predominates in the thigh and gluteal region and not in the ankles; c) the coexistence of a more or less permanent low back pain, which often persists in the decubitus, and d) the normality of the peripheral pulses. The picture is due to a usually subacute, slowly progressive compression of nerve roots,
Pseudocoma syndrome . Neurological syndrome characterized by immobility and lack of response to stimuli similar to those of coma, but with conservation of consciousness and produced by a virtually complete interruption of the efferent pathways of body expression.Its basic features are: a) it is a direct consequence of a cerebrovascular accident; b) neither spontaneously nor in response to stimuli, are there movements of the limbs of the trunk, the face or the pharynx, nor are sounds made, as occurs in coma, but with the particularity that blinking movements persist , and c) the individual is conscious, as evidenced by the examiner's blinking movements in direct response to questions. The syndrome is produced by thrombosis of the basilar artery in the ventral region of the pons, which interrupts all of the descending corticosterole and corticobulbar pathways, but without affecting the reticular activation system (consciousness) or the vertical motility pathways of the eye. (blink).
Pseudohypoparathyroidism syndrome . see Seabright-Bantam Syndrome
Pseudomiasthenic syndrome . see Eaton-Lambert syndrome
Cerebral pseudotumor syndrome . Syndrome characterized by neurological manifestations comparable to those of a brain tumor, but without detectable organic pathology.It predominates in young women and its features are the following: a) moderate, slowly progressive headache, occasionally accompanied by dizziness or mild diplopia, in patients with excellent general condition; b) bilateral papilledema associated with narrowing of the visual field and enlargement of the blind spot, in both cases of mild degree; c) hypertension of the cerebrospinal fluid, and d) normal or slightly decreased size of the cerebral ventricles, normality of tomographic and arteriographic studies, and absence of atypical elements in the analysis of cerebrospinal fluid. The condition is reversible, responds to corticosteroid treatment, and its cause is unknown.
Elastic pseudoxanthoma syndrome . see Groenblad-Strandberg syndrome .
Sézary syndrome . Syndrom of the leukemic phase of mycosis fungoides. It is observed in the advanced stages of mycosis fungoides (malignant cutaneous lymphoma), when neoplastic cells are already found in the circulating blood, and is defined by the presence of hyperpigmentation, dysmorphic plaques and diffuse thickening of the skin, and fundamentally, of a generalized erythroderma. In the terminal phases there is hepatosplenomegaly along with other manifestations of visceral invasion.
Shy-Dragger syndrome . Orthostatic hypotension syndrome with other neurological manifestations . The picture is that of idiopathic orthostatic hypotension syndrome, to which are added the manifestations of diffuse brain lesions. It is observed when degeneration of the extrapyramidal pathways, the basal ganglia, and the dorsal nucleus of the vagus predominates.
Sickert-Millikan syndrome . Syndrome of intermittent occlusion of the basilar artery. It is manifested by recurrent, transitory and more or less fleeting episodes of vertigo, loss of vision, diplopia, dysarthria, dysphagia, and sometimes hemiparesis.
Silvestrini-Corda syndrome . Hyperestrogenic syndrome of advanced chronic liver disease. It designates the set of feminization signs that are observed in cirrhosis, advanced liver, and that result from the inability of the liver to inactivate circulating estrogens: gynecomastia, eunucoid habit, loss of pubic and axillary hair, decreased libido, sterility and testicular atrophy.
Superior frontal silvian syndrome . see Motor Aphasia Syndrome
 Stein-Leventhal syndrome. Note the mammary hypoplasia, a tendency to obesity and virilization in a 16-year-old patient (Surós, Semiología Médica, 1968) |
Sipple syndrome . Type II syndrome of multiple endocrine neoplasms. It designates the presence of a pheochromocytoma (which is often bilateral and multiple), a medullary carcinoma of the thyroid, and a parathyroid hyperplasia. There may also be other tumors: meningiomas, glioblastomas, etc. The dominant symptoms are usually those of hyperparathyroidism, and less frequently those of pheochromocytoma. The disorder is inherited and is transmitted as an autosomal dominant trait.
Syringomyelia syndrome . Neurological syndrome of gliomatosis of the gray matter of the spinal cord. Syringomyelia is a proliferation of periependymal glia (gliomatosis) that destroys the medullary gray matter and affects mainly the cervical and lumbar segments. It is characterized, basically, by sensory disorders (syringomyelic dissociation, loss of thermal and pain sensitivity, with preservation of tactile and deep sensitivity), motor manifestations (spastic paralysis) and trophic alterations (syringomyelic arthropathy, atrophies, of the Aran-Duchenne type , etc.) The cause is unknown.
Sjögren's syndrome . Syndrome characterized by the triad of arthritis, xerophthalmia and xerostomia. It is a picture of dry eyes (xerophthalmia, keratoconjunctivitis sicca) and dry mouth (xerostomia) that occurs in individuals with rheumatoid arthritis. It has a chronic course and is attributed to autoimmune mechanisms.
Sluder syndrome . Sphenopalatine ganglion neuralgia syndrome. It is characterized by dull and continuous unilateral pain in the region of the upper jaw, which spreads to the interior of the neck and shoulder, and is accompanied by nasal obstruction, rhinorrhea and earache.
Sly syndrome . Mucopolysaccharidosis syndrome VII. It is a form similar to a mild Hurler syndrome and is characterized by: a) short stature with a keel chest; b) facial dysmorphia with flattened facies with coarse features, depressed nose and high subnasal sulcus; c) moderate mental retardation and d) striking inclusions in granulocytes. It is caused by a beta-glucuronidase deficiency and is transmitted as an autosomal recessive trait.
Spiller syndrome . Neurological syndrome of chronic spinal cord compression. It is a sequela of incompletely resolved pachymeningitis (remaining scars) and is manifested by pain and other sensory disorders, muscle weakness, ascending paralysis, and signs of transverse myelitis.
Steele-Rlchardson-Olszewski syndrome . Neurological syndrome of progressive supranuclear palsy. The onset is insidious and is characterized by alterations in balance, gait and unexpected falls, stiffness affecting mainly the neck and to a lesser extent the trunk muscles, difficulty in directing the gaze downward, and weakening of the volume of the voice. . In the final stage there is extreme rigidity, complete ophthalmoplegia, anarthria, and total disability.
Stein-Leventhal syndrome . Polycystic ovarian syndrome. It is seen in young women and is defined by the association of polycystic ovaries, oligomenorrhea, and sterility, which are accompanied by varying degrees of obesity, hirsutism, and breast hypoplasia. It is attributed to alterations in the synthesis of estrone.
Stevens-Johnson syndrome . Syndrome of a severe form of erythema multiforme. It is characterized by the development of ulcerative lesions located in the oronasal and anogenital mucosa, the eyes and viscera, with headache, arthralgia, fever and prostration. It can be fatal. There are idiopathic cases and others associated with viral diseases or ingestion of different drugs.
Still's syndrome . Syndrome of one of the forms of rheumatoid arthritis. It refers to a picture of chronic polyarthritis accompanied by generalized adenopathies, recurrent febrile episodes with skin rash, and splenomegaly; there may be pericarditis, pleurisy, and pneumonitis. There is anemia, leukocytosis, and negative tests for rheumatoid factor and antinuclear antibodies. Manifestations begin in childhood or adolescence and the cause is not precisely known.
Stokes-Adams syndrome . Syndrome associated with atrioventricular block. It is characterized by sudden loss of consciousness, with or without seizures, due to failure of the lower pacemaker in complete AV block.
Strachan syndrome . Syndrome characterized by neurological disorders and cutaneomucosal alterations of nutritional origin. It defines a picture of progressive development of eye disorders (decreased visual capacity, blindness), signs of peripheral neuropathy (paresthesia, ataxia, loss of superficial and deep sensitivity) and skin and mucous lesions (glossitis, stomatitis, etc.), It is observed in human populations that are malnourished or subjected to forced conditions of food restriction (eg, in besieged cities).
Sturge-Weber-Dimitri syndrome . Encephalotrigeminal angiomatosis syndrome. It is defined as the association of a flat facial angioma, of the capillary type and located in the trigeminal distribution territory, with a venous meningeal angioma, which is located more frequently in the parieto-occipital region. Clinically, it is manifested by extremely frequent epileptic seizures, sometimes with hemiparesis and hemianopia, and by mental retardation, while skull radiographs show the presence of characteristic linear calcifications.
Sudeck syndrome . Circumscribed post-traumatic osteoporosis syndrome. It designates the foci of localized osteoporosis, accompanied by pain and atrophy of the soft tissues, which sometimes develop as a consequence of small repeated trauma (eg, to the toes), and more often around the areas of fracture during the immobilization period.
Sweet syndrome . Acute febrile neutrophilia dermatosis syndrome. It is characterized by a non-pruritic eruption of red, painful, partly confluent nodules or plaques, which begins on the arms and usually affects women; there is high fever and neutrophilia. The cause is unknown.
Tabetic syndrome . Neurological syndrome of advanced syphilis. Tabes dorsalis is a posterior meningoradiculitis that leads to rapid sclerosis of the posterior cords of the spinal cord. Its basic manifestations are: a) ataxia with heel gait and muscular hypotonia; b) dissociation of sensitivity of the tabetic type (loss of deep and tactile sensitivity, with preservation of sensitivity to pain and heat); c) trophic alterations (bad plantar perforation, tabetic arthropathies), and d) positive serology for syphilis.
Takayasu syndrome . Syndrome of progressive occlusion of the branches of the aortic arch. It is characterized by a loss of pulse in both arms and in the carotids, together with symptoms caused by cerebral ischemia (syncope, transient hemiplegia, etc.), ocular (transient blindness, retinal atrophy, etc.), of the face (muscle atrophy) and arms (claudication). It is due to inflammation and progressive obliteration of the brachiocephalic trunk and the left subclavian and primitive carotid arteries, above their origin in the aortic arch. An autoimmune etiology is assumed.
 Still's syndrome |
Tapia syndrome . Neurological syndrome of injury to two nuclei of cranial nerves. It manifests as unilateral paralysis of the larynx (nucleus ambiguus) and tongue (nucleus of the hypoglossal), without paralysis of the soft palate.
Repetitive paroxysmal tachycardia syndrome . see Papp-Parkinson syndrome .
TAR syndrome . see Thrombocytopenic syndrome with absence of radius
Thalamic syndrome . Thalamus optic injury syndrome.Depending on which regions are predominantly affected, the syndrome includes variable combinations of some of the following manifestations that are observed on the side of the body, opposite to that of the lesion: a) total deep hemianesthesia with less marked superficial hemianesthesia; b) unpleasant and intense pain (thalamic pain) on the affected side, which appears after the acute period and in cases in which the posteroinferior nucleus was affected; c) motor alterations, including transient hemiparesis, reduction of voluntary motility and the appearance of abnormal movements (tremor and ataxia, synkinesias of imitation of choreic and athetosic movements); d) homonymous hemianopia, in posterior lesions, and e) neuropsychic manifestations, with marked reduction in language, verbal perseveration and ideas, and so on.
Taussig-Bing syndrome . Syndrome of a congenital heart disease with complex malformations. It is defined by: a) an anatomical triad made up of transposition of the great vessels, ventricular septal defect and thrust of the pulmonary artery, which produce secondary hypertrophy of the right ventricle; b) an oxygen saturation, which is greater in the blood of the pulmonary artery than in the blood of the aorta, and c) its clinical manifestations: pulmonary hypertension, permanent cyanosis, growth retardation, drumstick fingers, etc. .
Thieberge-Weissenbach syndrome . Scleroderma syndrome with diffuse calcifications. It is defined by the association of generalized scleroderma with diffuse calcinosis, which is attributed to concomitant parathyroid dysfunction.
Thomson's syndrome . see Rothmund-Thomson syndrome .
Anterior tibial syndrome . Tibial muscle necrosis syndrome. It manifests as rapid swelling, increasing tension, pain, and ischemic necrosis of the anterior tibial muscles. The skin becomes engorged and shiny, erythematous, and shiny as necrosis progresses. The cause is unknown, although it is supposed to be related to excessive exercise.
Tietze syndrome . Painful syndrome located in the chondrocostal joints. It clearly predominates in women between 35 and 50 years old, and is characterized by localized pain in one of the chondrosternal joints, and less frequently, in a sternoclavicular joint
Tolosa-Hunt syndrome . Unilateral pain syndrome of the face. It is characterized by unilateral facial pain, clearly predominant in the orbital region (first trigeminal branch), accompanied by ophthalmoplegia and uneven pupils. It is caused by inflammatory or tumor processes located in the vicinity of the superior orbital cleft or the cavernous sinus.
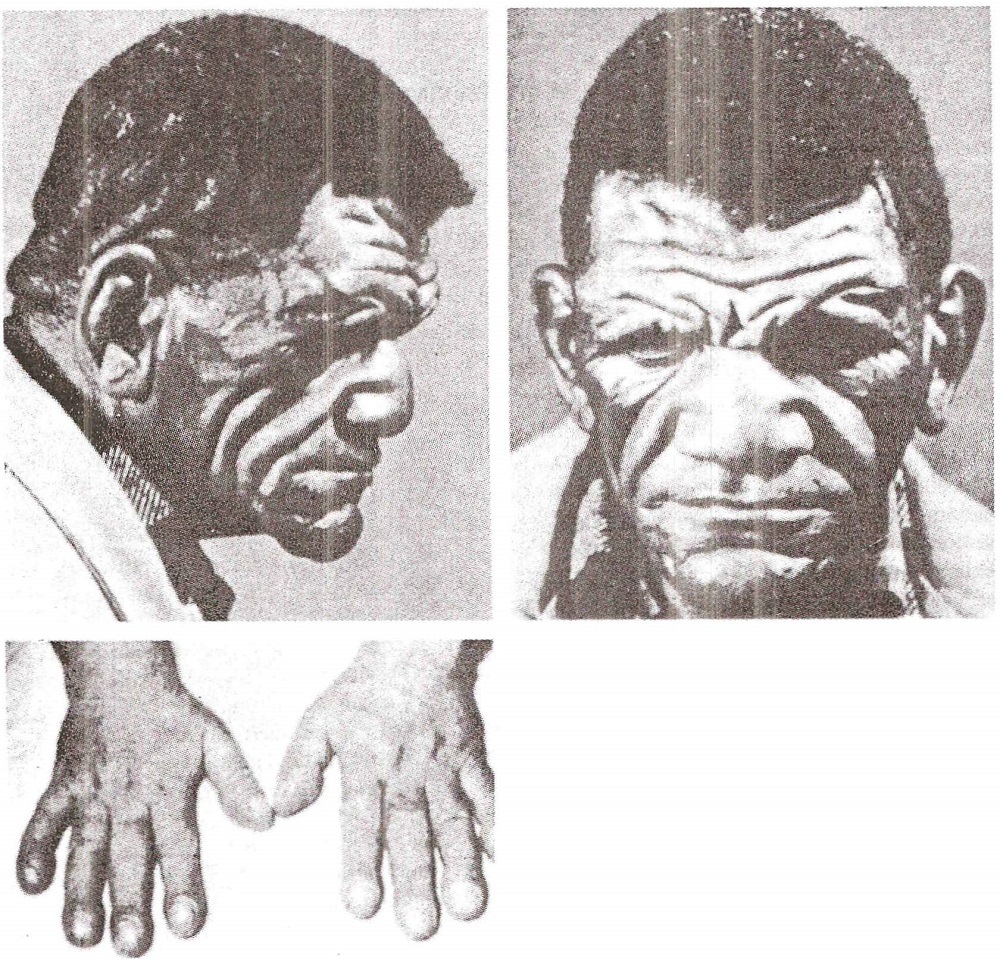 Touraine-Solente-Golé syndrome. Note the marked skin thickening and soft tissue hyperplasia of the fingers with curved nails (Modero Medicine, 1973) |
Toni-Fanconi syndrome . see Fanconi syndrome .
Touraine-Solente-Golé syndrome . Pachydermoperiostosis syndrome. It consists of a marked thickening of the skin of the face, forehead and scalp, with the formation of thick folds, periosteal neoformation and development of drumstick fingers. It is inherited as an autosomal dominant trait and usually appears around puberty.
Treacher-Collins syndrome . Syndrome of a hereditary picture with predominantly maxillofacial alterations. Its basic features are: a) bilateral hypoplasia of the lower jaw and malar bones; b) eyelid malformations, which include the presence of a coloboma in the outer third of the lower eyelid, and c) abnormalities of the auricle, sometimes of the middle and inner ear, and eventually deafness. It is a family disorder in which malformations occur during intrauterine life.
Trichorrinophalangeal syndrome . Syndrome characterized by skeletal, hair, and nose abnormalities . It is characterized by brachydactyly and axial deviation of the fingers, scoliosis or lordosis, short stature, sparse and thin hair, and a pyriform nose with a long subnasal groove. The cause is unknown and is compatible with an otherwise normal life.
Triple X syndrome . Hereditary syndrome of individuals with mental retardation and amenorrhea. It occurs in women and is defined as the association of mental weakness of variable degree, secondary amenorrhea, and unusual bodily abnormalities. Affected people have a trisomy of the sex chromosome X.
Trisomy syndrome 21 . see Down Syndrome .
Thrombocytopenic syndrome with absence of radius . Syndrome of a combined picture of cardiac and bone malformations , and hematological alterations. It is defined by the presence of a tetralogy of Fallot or defects in the interatrial septum, accompanied by thrombocytopenia and radial aplasia. It is a hereditary condition.
Capsular thrombosis syndrome . Neurological syndrome due to unilateral injury to the internal capsule. It is the classic syndrome, complete with alternating hemiplegia, in which there is paralysis of the half of the body and of the face on the opposite side to the brain injury. It is produced by the occlusion of the perforating branches of the middle cerebral artery.
Trousseau syndrome . Thrombotic syndrome associated with the presence of a neoplastic process. Appearance of spontaneous venous thrombosis, usually repeated, without apparent cause and predominantly in the lower limbs associated with the presence of an occult carcinoma. It is considered as a paraneoplastic syndrome.
Quadrigeminal tuber syndrome . Neurological syndrome of the unilateral lesion of the quadrigeminal tubercles. Its manifestations are basically auditory and ocular hearing loss of the central type accompanied by paralysis of the muscles dependent on the third cranial nerve, paralysis of the vertical gaze upward, paresis of convergence, nystagmus, and anisocoria with the Argyll-Robenson sign. The cause is usually tumor.
Carpal tunnel syndrome . Peripheral neurological syndrome due to compression of the median nerve. It is characterized by pain and tingling or burning paresthesias in the hand and fingers, which can cause degeneration of nerve fibers and lead to muscle atrophy. It results from compression of the median nerve in the carpal canal and is often bilateral.
Tarsal tunnel syndrome . Peripheral neurological syndrome due to compression of the posterior tibial nerve. It is due to the compression of this nerve in the tarsal canal, or its plantar branches, and is manifested by pain, numbness and paresthesia in the sole of the foot.
Turner syndrome . Syndrome characterized by gonadal agenesis and multiple anomalies of genetic cause (X chromosome monosomy , type XO). Its features are: a) short stature and short neck; b) lymphedema of the upper and lower limbs; c) gonadal agenesis with sexual infantilism, female external genitalia, mammary hypoplasia and primary amenorrhea (in women), and d) decreased blood estrogens with increased gonadotropins in the urine. The study of sex chromatin reveals that some of these patients are genetically "male" and others are genetically "female", despite the fact that there are no phenotypically differences between them.
Male Turner syndrome . see Noonan Syndrome
Uehlinger syndrome . Syndrome characterized by the association of generalized hyperostosis with localized thickening of the skin. The picture is defined by the development of a diffuse osteosc1erosis that affects the epiphyses, metaphyses, and diaphyses, and that is manifested by subperiosteal deposition of abnormal amounts of cancellous bone due to pain, swelling and stiffness of the joints and by marked thickening of the skin of the lower arm, and the forearm. It begins in puberty or adolescence and the cause is unknown.
Nail-patella syndrome . Hereditary syndrome characterized by bone and nail abnormalities and kidney disease. Its features are: a) wedge-shaped elbows, rudimentary kneecaps and horn configuration of the iliac bones; b) deformed nails with multiple clefts and c) signs of moderate chronic glomerulonephritis, usually of benign evolution. The picture is of an osteosdysplasia, of unknown cause, and is transmitted as an autosomal dominant trait.
Urbach-Wiethe syndrome . Lipoid proteinosis syndrome. The picture is due to the extracellular deposit of hyaline material in the skin and in the mucosa of the pharynx and larynx, and is manifested by: a) dysphonia or aphonia, macroglossia and dental anomalies; b) progressive development of skin papules of generalized distribution, yellowish-white in color and made up of intensely positive material in the PAS reaction, and c) presence of symmetrical calcifications in the area of the sella turcica. It is a granulomatous lipid storage disease that does not usually prevent a long life, and its cause is unknown.
Uremic-hemolytic syndrome . Infant acute renal failure syndrome associated with hemolysis, coagulation disorders and hemorrhages. It is a serious condition of early childhood and is characterized by the presence of intravascular hemolysis, thrombocytopenia, skin and mucosal hemorrhages and neurological manifestations, with intravascular coagulation phenomena and acute renal failure. The cause has not been established with precision, but it is attributed to an atypical reaction of the body in response to different virus infections.
Rapid emptying syndrome . Gastrectomy syndrome . Appearance of postprandial episodes of nausea, perspiration, palpitations, marked weakness, a sensation of heat in waves and sometimes diarrheal cramps, in individuals undergoing a partial gastrectomy with gastrojejunostomy. It is attributed to an excessively rapid emptying of gastric contents in the jejunum.
Vagal myocardial infarction syndrome . Syndrome of the infarcts of the inferior face of the heart. The picture includes functional digestive disorders (hiccups, belching, nausea or vomiting), vasomotor phenomena (paleness, sweating, hypotension, lipothymia) and heart rhythm disturbances (sinus or nodal bradycardia, blockage, sinoatrial, atrioventricular dissociation with interferences); in patients with myocardial infarction of the inferior face. It is seen relatively frequently and responds rapidly to atropine administration.
Van Buchem syndrome . Generalized cortical hyperostosis syndrome. Hereditary condition characterized by osteosclerosis of the skull, lower jaw, clavicles, ribs, and diaphysis of long bones, with increased serum alkaline phosphatase. Due to the progressive compression by the bone, optic atrophy, facial paralysis and perception deafness usually occur. It is transmitted as an autosomal, recessive trait.
Inferior vena cava syndrome . Inferior vena cava obstruction syndrome. Progressive edema of the lower extremities and scrotum, dilation of the superficial veins of the legs and the lower abdomen, oliguria and sometimes ascites. It is seen in severe conditions such as generalized peritonitis and ascending thrombosis of the veins of the lower limbs and is usually a terminal complication.
Superior vena cava syndrome . Superior vena cava flow obstruction syndrome. It is characterized by edema of the face, neck, upper chest, and upper limbs (edema edema), fixed engorgement of the neck veins, predominantly facial cyanosis, and painful hepatomegaly. It is due to extrinsic compression of the vessel walls, usually due to a tumor.
Verner-Morrison syndrome . see WDHA Syndrome .
Vernet syndrome . Neurological syndrome of the base of the skull. It is characterized by progressive and simultaneous unilateral palsy, although of variable degree, of the IX, X and XI cranial nerves, and is due to compression exerted by neurinomas, sarcoidosis foci, or infiltrating tumors of the skull base.
Crag vertex syndrome . see Gradenigo syndrome .
Cervical vertigo syndrome . Neurological syndrome due to insufficiency of the vertebral circulation. Sudden, transient episodes of dizziness, vertigo, nystagmus, ataxia, and diplopia, or combinations of some of these symptoms, which are usually triggered by forced or overly wide movements of the head and neck. It is caused by compression or spasm of the vertebral artery
Villaret syndrome . Syndrome of injury to cranial nerves IX, X, XI and XII, and of the cervical sympathetic, due to tumor compression. It is unilateral and is characterized by: a) loss of taste in the posterior third of the tongue (lesion of the IX cranial nerve); b) paralysis of the superior constrictor of the pharynx, with difficulty in swallowing solids (X nerve); c) paralysis of the soft palate and the jaws, with associated anesthesia (X nerve); d) paralysis of the vocal cord (X nerve); e) paralysis of the trapezius and sternocleidomastoid (XI nerve); f) paralysis of the middle of the tongue (XII nerve), and g) Horner syndrome (cervical sympathetic paralysis). It is produced by tumors of the parotid and carotid body, and tumor or tuberculous adenopathies.
Adrenal virilization syndrome in adults . Variety of adrenogenital syndrome. Its manifestations, in women, are those of excessive androgen production: hirsutism, acne, seborrhea, temporary baldness, increased muscle mass and strength, amenorrhea with uterine atrophy and hypertrophy of the clitoris, masculinization of the voice, etc. . In the adult male, obviously, the picture is more difficult to detect. The cause is often tumorous (adrenal adenomas or carcinomas) and there is a marked increase in urinary 17-ketosteroid excretion.
Vogt syndrome . Caudate nucleus and putamen injury syndrome. It is characterized by the presence of athetosic movements, rhythmic oscillation of the limbs, and spasmodic and explosive crying and laughter. There is no paralysis or sensitive phenomena.
Vogt-Koyanagi syndrome . Syndrome characterized by alterations in the skin and the sense organs. It is an association of vitiligo and areas of alopecia, with eye disorders and deafness, sometimes accompanied by labyrinthitis.
Volkmann syndrome . Necrosis and muscle retraction syndrome due to acute regional ischemia. It is an ischemic condition, due to interruption of the arterial flow directly related to the compression exerted by fracture foci, or by the casts made to immobilize them, which usually affects the hand and the flexor surface of the forearm and is manifested by intense pain, swelling, cyanosis , paresthesias and inability to move the fingers, and if there is no rapid resolution, due to muscle necrosis and scar retraction in flexion.
Waardenburg syndrome . Hereditary syndrome of individuals with deafness and predominantly facial abnormalities. Albinism of the eyelashes and a tuft of white hair in the frontal region, union of the eyebrows in the midline, heterochromia of the iris, a flattened nose and congenital deafness are observed. It is transmitted as an autosomal dominant trait.
Wallenberg syndrome . Neurological brainstem syndrome with sensory and cerebellar disorders and involvement of cranial nerves V, IX, X and XI.It is characterized, on the side of the injury, by corneal anesthesia and paralysis of the masticatory muscles (injury to the V nerve); Difficulty swallowing and loss of taste in the posterior third of the tongue (IX nerve); paralysis of the soft palate and vocal cord (X nerve); paralysis of the trapezius and sternocleidomastoid (Xl nerve); Horner syndrome (lesion of the descending pupilodilator pathway), and cerebellar ataxia (spinocerebellar and olivocerebellar pathways); and on the opposite side due to loss of thermal sensitivity and pain (lateral spinothalamic bundle). The picture is produced by the occlusion of the posteroinferior cerebellar artery.
Waterhouse-Friderichsen syndrome . Syndrome of the fulminant form of cerebrospinal meningitis. It is a picture of sudden onset and rapid evolution, characterized by paleness, dyspnea, cyanosis, high fever with chills, petechiae and ecchymosis on the skin and mucous membranes, collapse and coma. It is observed in pediatric patients and is the consequence of a very serious sepsis, usually due to meningococci or pneumococci.
WDHA syndrome . Syndrome of diarrhea-producing pancreatic tumors. The name derives from the initials; English of its characteristic manifestations: watery diarrhea (watery diarrhea), hypokalemia (hypokaliemia) and achlorhydric (achlorhydria). The diarrhea is usually profuse, severe enough to sometimes lead to shock, or to produce alarming hypokalemia; there is a decrease in basal gastric acid secretion, and hypercalcemia and hyperglycemia are often present. The condition is produced by non-insulin tumors of the islets of Langerhams and its manifestations are apparently due to the hypersecretion of vasoactive intestinal polypeptides.
Weber syndrome . Neurological brainstem syndrome with alternate hemiplegia and third cranial nerve involvement. The picture is that of a hemiplegia on the side opposite to that of the injury, with palpebral ptosis, strabismus, and loss of light reflexes and accommodation on the side of the injury. It is typical of lesions, almost always vascular, that affect the pyramidal bundle at the foot of the cerebral peduncle and the root fibers of the third cranial nerve.
Weber-Christian syndrome . Nonsuppurative nodular panniculitis syndrome. It is characterized by the formation of recurrent painful nodules in the subcutaneous fatty tissue, with fever and alteration of the general state; the nodules evolve leaving a central depression. The prognosis is benign and the cause is unknown.
Wegener syndrome . Necrotizing respiratory granulomatosis syndrome. It is characterized by necrotizing vasculitis and granulomatous infection of the respiratory tract and kidneys, and often of other organs. There is headache, fever, arthralgia, sinusitis, otitis, cough, hemoptysis, etc. It predominates in the middle age of life and is considered related to hypersensitivity phenomena.
Weill-Marchesani syndrome . Congenital mesodermal dystrophy syndrome. It is characterized by: a) short stature with brachycephaly and brachydactyly; b) wide thorax with masses; developed muscles that give the impression of great body strength; c) ocular alterations, with ectopia of the lens and often glaucoma and myopia; d) marked joint stiffness, and occasionally e) congenital pulmonary stenosis. It is transmitted as an autosomal recessive trait.
Werdnig-Hoffmann syndrome . Familial spinal muscular arthrophy syndrome . It is characterized by the early installation (early childhood) of hypotonia and muscle atrophy, followed by complete flaccid paralysis and finally death. It is due to a degeneration of the cells of the anterior horn of the medulla and is transmitted as an autosomal recessive trait.
Wermer syndrome . Multiple endocrine neoplasms syndrome type 1 .It is defined by the presence of tumors or hyperplasia of the parathyroid, thyroid, adrenal glands, the islets of Langerhans and the pituitary, and by a clinical picture that depends on the systems; hyperfunctional at the time the individual consults. Combinations of the following conditions can be observed: a) hypoglycemia; b) hypercalcemia with nephrocalcinosis; c) multiple peptic ulcers (Zollinger-Ellison syndrome); d) multiple lipomas of the skin; e) disorders produced by expansion of a pituitary tumor or by dysfunction of this gland (headache, gradual loss of vision, secondary amenorrhea, etc.), and less frequently, a Cushing syndrome, hepatomegaly and other manifestations.
Werner syndrome . Hereditary syndrome with skin changes and a tendency to develop malignant processes. It is characterized by premature aging, cataracts, scleroderma-like disorders, leg ulcerations, and atherosclerosis, with an increased tendency for the development of sarcomas and meningiomas.
Wernicke syndrome . Presbyophrenia syndrome. It is characterized by mental weakness with lack of fixation and disorientation, anxiety, hallucinations, delusions, and other disorders. It is a psychopathy typical of old age.
Weyers-Thier syndrome . Oculovertebral dysplasia syndrome. It is characterized by multiple ocular abnormalities (microphthalmia, anophthalmia, etc.), facial asymmetry, frontal hypoplasia and orbital dysplasia, and malformations of the spine and the insertion of the ribs. It is due to a disorder early in the development of the embryo and affects only males.
Wilfred Harris syndrome . Cranial nerve IX irritation syndrome. Intense attacks of paroxysmal pain located in the throat and sometimes radiating to the ear. It is repeated with variable periodicity, sometimes for no apparent reason and others; triggered by swallowing. The etiology is unknown, although from the clinical point of view the picture is in every way comparable to that of trigeminal neuralgia.
Wilson-Mikity syndrome . Clinical and radiological syndrome of pulmonary immaturity. It is the association of a persistent cyanosis in the newborn, with characteristic radiological manifestations: multiple foci of localized emphysema, 1 to 10 mm in diameter, surrounded by a condensation band. Other features are its presentation in premature infants, with very low birth weight, the absence of signs of infection, its slow course, high mortality, and the possibility of a complete recovery in those who survive.
Wolff-Parkinson-White syndrome . Electrocardiographic and clinical syndrome of abnormal atrioventricular excitation. It is defined by the presence, in the electrocardiogram of a normal P wave, a short PR (less than 0.11 sec) and a widened QRS with filling of its initial portion (delta wave) abnormality that is associated with a frank tendency to development of episodes of paroxysmal atrial tachycardia or atrial fibrillation. It is observed in young subjects without other signs of heart disease, and is due to the existence of accessory conduction pathways parallel to the bundle of His.
Woltman-Moersch syndrome . S índrome tetaniforme of noninfectious cause. It is observed in adulthood, without distinction between the sexes, and is characterized by: a) the appearance of intermittent spasmodic contractions, of irregular distribution, which predominate in the muscles of the trunk and extremities; b) parking for prolonged periods under these conditions, or slow progression with an increase in the affected muscle groups and longer duration of contractions, and c) in the latter case, evolution towards a state of virtual generalized rigidity. The cause is unknown, although it is attributed to an acquired deficiency of physiological mediators that inhibit motor neuron activity.
Wunderlich syndrome . Spontaneous perirenal hematoma syndrome. It is abruptly manifested by intense unilateral low back pain, signs of acute anemia and rapid appearance of a palpable lumbar tumor, in addition there is nausea and vomiting, meteorism due to reflex ileus, lumbar contracture and oliguria. It can appear in the course of kidney (pyelonephritis, tumors, etc.), hematic (hemophilia, leukemia) or vascular (periarteritis, caval thrombosis, etc.).
Zollinger-Ellison syndrome . Ulcerogenic pancreatic tumor syndrome. It is manifested by a characteristic triad: intractable, sometimes fulminant, peptic ulcers, extreme gastric hyperacidity, and watery diarrhea or steatorrhea. It is caused by the presence of non-beta tumors of the islets of Langerhans, single or multiple, benign or malignant, and is currently considered type I of multiple endocrine neoplasms.