
 English
English  Português
Português  Español
Español Fernando L. Soldano, Antonio Molina Rojas, Gustavo Lavenia
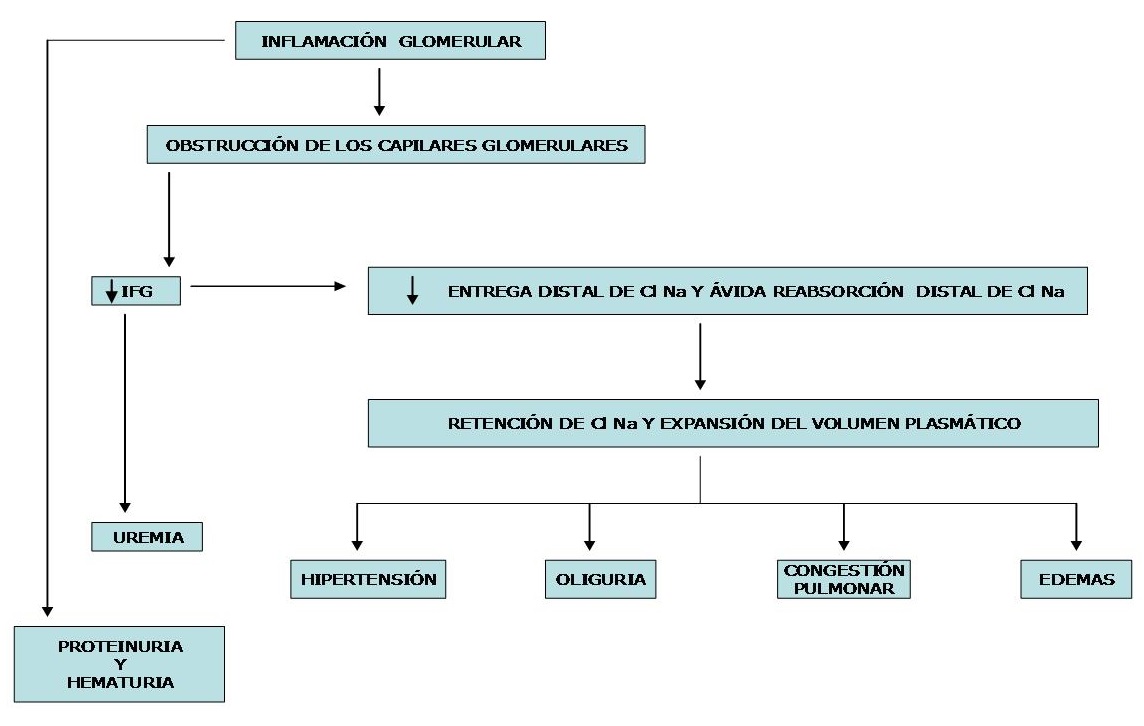
Syndrome characterized by macro or microscopic hematuria (capital sign), arterial hypertension (due to retention of H 2 O and sodium), proteinuria in the non-nephrotic range (with or without edema), and findings in the urinary sediment, of hematic casts. In addition, it can present clinically with renal failure, and occasionally oliguria. Acute nephritic syndrome is due to several morphological entities that manifest diffuse glomerular inflammation. Its presentation can be acute or chronic, in the acute form it generally occurs after infection of the upper airways or skin. In chronic forms, it has an insidious onset, with progressive impairment of kidney function, accompanied by proteinuria, hematuria, and arterial hypertension.
Hematuria : indicates glomerular damage, it can be asymptomatic, micro or macroscopic. The appearance of hematic casts suggests glomerular inflammation of any etiology. The finding of even 1 hematic cylinder is diagnostic of the syndrome.
Arterial Hypertension : the alteration of the filtering, retains sodium and water, causes expansion of plasma volume, raising blood pressure, as the first mechanism and is also caused by the activation of the renin-angiotensin-aldosterone system. The appearance of hypertensive encephalopathy usually occurs in the acute forms.
Proteinuria : present in almost all glomerulopathies, although to varying degrees. If it is mild, it is asymptomatic, in severe cases (> 3.5 g / 24 h.) It will be clinically evidenced with edema.
Edema : generalized or localized (eg periorbital and sacrum in decubitus position).
Renal Insufficiency : indicative of a severe fall in glomerular filtration rate, increased uremia and creatininemia.
Oliguria : It occurs in some acute forms and is characteristic of the rapidly evolving one, leading to a uremic syndrome.
The following diagram shows the evolution that leads to clinical manifestations in a patient with haematuria and abrupt-onset decrease in GFR.
Pathophysiology:
Most glomerulonephritis are immunologically mediated by:
a) Immune complexes (IC): an antigen that can be exogenous or endogenous binds to its antibody in the glomerulus or both are deposited there, after coupling in the circulation.
b) anti-glomerular basement membrane antibodies (anti-MBG): the antibody that is an endogenous IgG, binds to the basement membrane that is the antigen.
In both types of glomerulonephritis, the antigen-antibody reaction activates the complement components causing chemotaxis, cytolysis, and increased vascular permeability. Neutrophils release proteolytic enzymes and vasoactive platelets amines. Mesangial cells proliferate and lymphocytes cause cytolysis. The basement membrane tends to thicken, podocytes fuse, thrombi form, and acidophilic cellular material is deposited, obliterating the glomerulus. This hyalinization (sclerosis) is irreversible. Prostaglandins, leukotrienes, serotonin, histamine, angiotensin II, etc. participate in phlogosis.
The answers are variable: if few glomeruli are affected, GN is focal; if all or most are altered it is generalized. If the affected glomeruli are only partially, the GN is segmental; it is diffuse when they are in their entirety. Immunofluorescence detects immunoglobulins, complement components, fibrin, properdin, etc. When these manage to enter Bowman's space, they stimulate the migration and proliferation of monocytes and epithelial cells, forming crescents that cause the Rapidly Evolutionary GN picture.
Multiple experimental models for IC (Immunocomplexes) have been described that confirm the pathogenesis of Human GN. One is Serum Disease nephritis: a rabbit is injected with bovine albumin and between 8 to 14 days it presents joint, cardiac and kidney lesions. The animal has developed antibodies that form IC with bovine albumin, these are deposited in the glomerulus producing intense cell proliferation. The rabbit suffers from hypertension, proteinuria, hematuria and oliguria that tend to resolve spontaneously. Histological and clinical responses are variable, with acute, chronic, proliferative, exudative, membranous, and sclerosing lesions. The other experimental nephritis is Massugi's nephritis, in which a rabbit is injected with a rat kidney extract, resulting in the formation of anti-rat kidney antibodies. If the serum from this rabbit is now injected into another rat, it undergoes a GN (heterologous phase). Steblay demonstrated that the rabbit develops an anti-glomerular and pulmonary basal membrane IgG. In a second phase, the rat reacts by forming antibodies against rabbit Ig. This causes a second glomerulonephritic episode (autologous phase). In humans, the Good – Pasture syndrome of unknown etiology is similar to the Steblay model due to its GN and lung lesions. In some cases, Focal GN and in others there are severe pulmonary hemorrhages and crescents that plug Bowman's space, causing oligoanuria. the rat reacts by forming antibodies against rabbit Ig. This causes a second glomerulonephritic episode (autologous phase). In humans, the Good – Pasture syndrome of unknown etiology is similar to the Steblay model due to its GN and lung lesions. In some cases, Focal GN and in others there are severe pulmonary hemorrhages and crescents that plug Bowman's space, causing oligoanuria. the rat reacts by forming antibodies against rabbit Ig. This causes a second glomerulonephritic episode (autologous phase). In humans, the Good – Pasture syndrome of unknown etiology is similar to the Steblay model due to its GN and lung lesions. In some cases, Focal GN and in others there are severe pulmonary hemorrhages and crescents that plug Bowman's space, causing oligoanuria.
|
Causes of Nephritic Syndrome
|
Primary Glomerulonephritis:
a- IgA mesangial nephropathy (IgA):
Named for the discovery of IgA as a deposit. IgAN is the most common primary glomerulonephritis in most of the world, but with some geographic differences. The kidney disease detection campaigns and the kidney biopsy policy largely explain these epidemiological differences. For unknown reasons it is very rare in the black race. In adults, it is the leading cause of biopsied kidney disease. IgA nephropathies can be primary and secondary. In the primary ones, the idiopathic form (Berger's disease) is found without systemic manifestations and with systemic manifestations (Scholein-Henoch purpura). Among the secondary forms, it can be related to autoimmune diseases (SLE, Reiter's disease, Psoriasis, RA, Wegener, Mixed Cryoglobulinemia, etc.), infectious diseases (HIV, TBC,
Clinic
It is more common in males, who constitute about 74% of all cases. 80% of cases are diagnosed between 15 and 65 years of age (adults). In the Pathological Anatomy, the OM presents an increase in mesangial matrix and mesangial hypercellularity.
The most common clinical forms are the following: asymptomatic urinary disorders: That is, microhematuria associated with proteinuria no greater than 1 gr / 24 hours. Also gross hematuria, nephrotic syndrome and rarely acute renal failure.
Diagnosis
The diagnosis should be suspected in the face of the most typical pictures (urinary abnormalities or isolated or recurrent hematuria), with a normal urological study (radiographs, renal ultrasound, and cystoscopy), in the absence of urinary infection.
b- Mesangial proliferative glomerulonephritis:
It is characterized by presenting an increase in the mesangial matrix and mesangial hypercellularity with normal capillary wall and variable mesangial dense deposits on light microscopy. They are classified according to immunofluorescence according to the predominance of deposits, in GN by IgM, GN by IgG, GN by C3, GN by C1q, or deposits may not be found and only proliferation. They can take focal or diffuse forms and there are primary and secondary varieties (LES, AR). The most common clinical forms are the following: asymptomatic urinary disorders: that is, microhematuria associated with proteinuria of no more than 1 gr / 24 hours. Also gross hematuria, nephrotic syndrome and rarely acute renal failure.
c- Mesangiocapillary glomerulonephritis (Membranoproliferative):
Among all glomerulonephritis, mesangiocapillary is one of the least frequent. Classically mesangiocapillary glomerulonephritis (GNMC) has been classified as idiopathic and secondary, the former being subdivided into types 1, 2 and 3 according to histology. It receives different names, also called, membranoproliferative, hypocomplementémic or lobular.
Pathological anatomy:
The common characteristic of the different types of CMNG is an increase in mesangial matrix, proliferation of mesangial cells, and thickening of the capillary wall.
Here we describe the most typical histological findings of the three types of glomerulonephritis:
GNMC Type 1:
Mesangiocapillary glomerulonephritis with subendothelial or classic deposits
Optical microscopy:
The most common histologic feature is endocapillary hypercellularity and diffuse thickening of the capillary wall. This hypercellularity is due to both the presence of mesangial cells and leukocytes, mainly neutrophils and, to a lesser extent, eosinophils. The thickening of the wall is due to the interposition of mesangial and polymorphonuclear matrix, which gives the wall an appearance of double contour. In addition, there is a decrease in capillary lumen with the presence of "hyaline thrombi", which are not true thrombi but aggregates of immune complexes.
GNMC focal
It is a variant of type 1, where the aforementioned alterations appear only in some glomeruli and its prognosis is relatively favorable.
GNMC Type 2
Mesangiocapillary glomerulonephritis with basement membrane deposits or dense deposit disease
Optical microscopy:
As in type 1, mesangial proliferation, an increase in the mesangial matrix and thickening of the basement membrane are observed, but to a more variable degree, being able to simulate other types of glomerulonephritis, hence electron microscopy and immunofluorescence play an essential role. in your diagnosis. The typical lesion is a structural alteration of the basement membrane at the expense of a densoelectronic material of unknown composition that is not always visible in the optics, so to diagnose this nephritis, observation in electron microscopy is required.
GNMC Type 3:
Mixed mesangiocapillary glomerulonephritis (membranous and proliferative)
It is a variant of type 1, in which changes are observed in the capillary wall similar to those of the membranous. Immunofluorescence shows granular deposits of C3, IgG and IgM, especially in the capillary wall.
Clinic
GNMC can present with any clinical syndrome of glomerular disease, the most frequent being asymptomatic urinary anomalies (non-nephrotic proteinuria and microhematuria) and nephrotic syndrome, which account for 30% and 50%, respectively. Mesangiocapillary GN affects all age groups, although it is more frequent between 5-30 years, being exceptional from the seventh decade of life. According to the histological type, there are distinctive clinical characteristics: type 1 is associated with infections with hypocomplementemia and a mean age of onset of around 24 years. Type 2 affects younger patients with a higher incidence of acute renal failure and rapidly progressive nephritis. Type 3 is frequently asymptomatic, being more frequent the finding of microhematuria and proteinuria.
d- Extracapillary glomerulonephritis:
Glomerulonephritis with extracapillary proliferation is characterized by the existence of an accumulation of cells in the form of "crescents" that displace and occupy the normal structures of the glomerular tang. This proliferation is composed of cells with an epithelial appearance, located in the central area of the glomerular tang, a reason that justifies the name of extracapillary proliferation. These crescents are made up of several types of cells, most notably macrophages, which have passed into Bowman's space through the ruptured glomerular capillary wall, and epithelial cells in the parietal layer of Bowman's capsule. The initial lesion that triggers the formation of crescents lies in the rupture of the glomerular basement membrane. This rupture allows the passage of fibrin and monocytes into Bowman's space. In synthesis, the pathogenic sequence would be the following: 1) deposit of antibodies, immune complexes and complement that damage the capillary wall, in a process in which mononuclear cells and cellular immunity seem to play a fundamental role, 2) passage of fibronectin and of fibrin to the urinary space, 3) attraction of circulating monocytes, 4) proliferation of epithelial cells of Bowman's capsule, 5) increased synthesis of matrix proteins with formation of crescents, and 6) evolution towards fibrosis, due to increased collagen synthesis and fibroblast infiltration.
Rapidly progressive glomerulonephritis defines, in clinical terms, a group of glomerular diseases characterized by: 1) the presence of crescents in more than 50% of the glomeruli, excluding the completely sclerosed ones, and 2) a clinical course characterized by progressive deterioration and rapid renal function, such that, in the absence of treatment, about 85% of patients reach end-stage renal failure within a few days, weeks or months. When the percentage of crescents is less than 50%, the evolution is more favorable and, if not, not attributable to extracapillary proliferation. Other entities that have a similar clinical picture are: some forms of acute tubular necrosis, acute immunoallergic interstitial nephritis, hemolytic uremic syndrome, atheroembolic disease and myeloma kidney.
|
Classification of extracapillary glomerulonephritis
|
Clinic
It appears in middle-aged or adult patients, and equally in both sexes. Goodpasture syndrome is more common in young men. Sometimes there are arthralgias, myalgias, or a flu syndrome. Some environmental factors are triggers for the entire clinical picture, such as tobacco, cocaine, respiratory infection, pulmonary edema, and exposure to hydrocarbons. Renal involvement is characterized by oliguria, hematuria, or both. Blood pressure is normal or slightly elevated. Occasionally, there is asymptomatic alveolar hemorrhage that is diagnosed after the finding of macrophages loaded with hemosiderin in the sputum or by increased pulmonary uptake of CO. As a laboratory finding, proteinuria is not usually nephrotic and hematuria of glomerular origin is practically constant. Glomerular filtration progressively decreases very rapidly. The serum complement is normal. Iron deficiency anemia occurs in cases of alveolar hemorrhage. 95% of patients have elevated levels of IgG α-MBG antibodies, detected with RIA or ELISA. Some antigens of the major histocompatibility system are more common than in the healthy population (HLA-DR2, HLA-DR4, and others).
Postinfectious Secondary Glomerulonephritis:
Acute endocapillary glomerulonephritis.
Endocapillary glomerulonephritis is characterized by diffuse cellular growth in the glomerular tang. Glomerular hypercellularity is due to proliferation, especially of mesangial cells, and to infiltration of inflammatory cells. The clinical presentation is variable. The classic presentation is acute nephritic syndrome, but it can sometimes manifest only with microscopic hematuria and varying degrees of proteinuria; more rarely, it can present with nephrotic syndrome. Physiopathologically, there is an endogenous or exogenous antigenic stimulus and an immune response that takes the renal glomerulus as the shock organ. Depending on the etiology, the complement system is activated. In addition, there is a local activation of the coagulation cascade.
Acute post-streptococcal glomerulonephritis is the typical example of acute endocapillary glomerulonephritis.
Acute post-streptococcal glomerulonephritis (GNAPE)
The clinical picture manifests itself 10 to 15 days after a red sore throat, due to streptococcus serotype M 1, 3, 4, 6, 12, 25 and 49 or after a Pyoderma due to serotype M 2, 31, 49, 51 , 52, 55, 56, 57, 59, 60 and 61. The lesion is proliferative and diffuse, with abundant neutrophils.
The clinical manifestations are hematuria, edema, hypertension, oliguria, weakness, anorexia, and occasionally dull pain in the lumbar fossae. The complete clinical picture of acute nephritic syndrome (hematuria, edema, and arterial hypertension) occurs in 50% of cases of symptomatic GNAPE, but some of these manifestations occur in 95% of clinical cases. Thus, the terms acute glomerulonephritis and nephritic syndrome are sometimes used interchangeably. Less than 4% of patients have proteinuria in the nephrotic range (> 3.5 g / day) and <1% have a rapidly progressive elevation of nitrogen products.
It commonly improves spontaneously, patients in whom proteinuria persists or recurs, progress to chronic renal failure.
Secondary Multisystemic Glomerulonephritis:
Alport Hereditary Glomerulonephritis:
It is an autosomal dominant disease associated with deafness and eye disorders. Glomerulonephritis is focal and proliferative with interstitial inflammation, fibrosis, and foam cells. The highest percentage of appearance is between 5 and 20 years of age and clinically it presents with hematuria, proteinuria, hypertension and occasionally nephrotic syndrome.
Lupus Glomerulonephritis:
It presents clinically with a picture of fatigue, generalized weakness, anorexia, weight loss, fever, arthralgia, malar erythema, dermatosis and alopecia. The glomerulus may show HF deposits with normal, focal or generalized proliferative, membranous, or rapidly evolving histology.
Systemic Vasculitis:
They constitute a group of clinical entities that affect (inflammation and necrosis predominantly a type and size of vessel and organs. Among them are:
- Wegener's granulomatosis: Causes focal or rapidly evolving GN, presents with upper and lower air tract involvement such as epistaxis, rhinorrhea, sinusitis and deformation of the nasal septum, pulmonary cavitation, cough, dyspnea and general manifestations (general syndrome): such as low-grade fever , general decline, anorexia and weight loss.
- Panarteritis nodosa (PAN): Medium and small arteries are affected, essentially the kidney, heart and central nervous system, it presents with a general syndrome and usually respects the lung, Cardiac involvement (70%) in the form of angina, infarction or pericarditis. Renal involvement (80%) with proteinuria, hematuria, cell casts and progressive IR.
- Microscopic PAN: affects small arteries, is associated with Wegener's G. and idiomatic necrotizing glomerulonephritis. Clinically it presents with rapidly evolving GN.
- Schonlein-Henoch glomerulonephritis: It is focal or rapidly evolving. It is associated with purpura, arthritis, and abdominal cramps.
- Churg-Strauss disease: allergic granulomatosis and vasculitis, presents allergic manifestations, rhinitis, asthma and typically eosinophilia in peripheral blood. 70% present cutaneous manifestations (subcutaneous nodules on the extremities, palpable purpura, maculopapular rash and petechiae). Renal involvement with focal and segmental GN.
Essential mixed cryoglobulinemia:
GN can be proliferative or membranous and can be associated with purpura, arthralgias, and Raynaud's phenomena.
Microangiopathic Hemolytic Anemias: HUS (Hemolytic Uremic Syndrome) and TTP (Thrombocytopenic Thrombotic Purpura)
They are different entities, which share common pathogenic mechanisms and clinical manifestations. They present with microangiopathic hemolytic anemia, thrombocytopenia, and ARF. HUS is more frequent in children, preceded by an episode of diarrhea, between 40% and 80% have ARF and 90% have hematuria and proteinuria. TTP, is more frequent in adults, in the third and fourth decade of life, is usually accompanied by fever and neurological manifestations.