
 English
English  Português
Português  Español
Español Fernando L. Soldano, Antonio Molina Rojas, Gustavo Lavenia
Syndrome characterized by proteinuria (capital sign), edema, dysproteinemia (hypoalbuminemia, hypergammaglobulinemia), and dyslipidaemia. It is the clinical consequence of the increased permeability of the glomerular capillary wall, therefore, it is a manifestation of glomerular kidney disease. Proteinuria is considered to cause nephrotic syndrome if it is greater than 3.5 g / 24 h / 1.73 m2 in adults or 40 mg / h / m2 in children (nephrotic range proteinuria). However, the clinical manifestations of nephrotic syndrome may not appear with figures higher than this value or be seen with figures lower than 3.5 g / 24 h. The importance of nephrotic syndrome lies in the magnitude of proteinuria, and in the underlying glomerular lesion, therefore,
Pathophysiology of proteinuria in nephrotic syndrome
The central pathophysiology of nephrotic syndrome lies in increased glomerular permeability and the consequent loss of protein in the urine. The rest of the alterations described in the nephrotic syndrome are a direct consequence of this massive proteinuria.
The normal glomerular capillary wall, composed of endothelial cells, glomerular basement membrane, and visceral epithelial cells, constitutes a barrier due to size and electrical charge to the passage of proteins larger than 70 kd. In nephrotic syndrome, proteinuria could be caused, at least theoretically, by an electrochemical disorder (loss of electronegativity of the filtration barrier), or by a structural alteration of the filtration membrane that conditions an increase in the size of the pores Of the same. In size of the pores in the wall of visceral epithelial cells, it plays a fundamental role, since they would prevent the passage of macromolecules larger than 150 kd. Various adhesion molecules, such as nephrin, as well as integrins and proteins of the cytoskeleton, appear to be fundamental constituents of these pores. In addition, some theoretical models suggest the existence of other pores with a larger radius, whose number would be increased in glomerular damage, favoring massive proteinuria. These pores would shrink with antiproteinuric drugs such as angiotensin converting enzyme (ACE) inhibitors. The structural alteration of glomerular epithelial cells is characteristic of all the processes associated with nephrotic syndrome. The limited proliferative capacity of these cells after noxa would favor proteinuria and plasma protein insudation, which by light microscopy would translate into protein deposits, alterations in the glomerular basement membrane and glomerular mesangium abnormalities.
The charge-dependent barrier is due to the negative charges of the polyanionic glycosaminoglycans rich in heparan sulfate of the glomerular basement membrane, which would restrict the passage of small plasma polyanionic proteins with a size between 70-150 kd, mainly albumin. For this reason, positively charged proteins, at equal sizes, present a greater renal clearance than negatively charged proteins. Minimal change nephropathy is the paradigm of diseases caused by an electrochemical glomerular disorder. In these cases, no morphological alterations are observed with light microscopy, and proteinuria is very selective (albumin and other negative proteins are lost, while those of higher molecular weight, such as IgG, are retained).
Small modifications in the permeability properties of the glomerular wall produce significant losses of proteins of intermediate molecular weight (between 40 and 150 kd). These include: albumin, IgG, transferrin, ceruloplasmin, and glycoprotein. Small amounts of slightly larger proteins such as small forms of HDL (200 kd) are also lost. Very high molecular weight proteins such as IgM, macroglobulins, fibrinogen, factor XIII, fibronectin, and larger lipoproteins are not lost, even with large alterations in glomerular permeability and selectivity. From a practical point of view, proteins larger than 200 kd are not lost in nephrotic syndrome.
Regardless of the extent of glomerular damage, other factors can condition a very wide variation in proteinuria, such as glomerular filtration, renal plasma flow, activity of the renin-angiotensin system, plasma production and concentration of albumin, protein intake daily and the administration of antihypertensive drugs.
Nephrotic Syndrome Clinic
Hypoalbuminemia
Albumin is the most abundant plasma protein. Filtered albumin is catabolized in part by the renal tubule, the catabolic rate of which increases and may represent up to 20% of the filtered albumin in nephrotic syndrome.
To compensate for the losses, the liver increases the rate of albumin synthesis by up to 300% through transcriptional mechanisms. This increase is correlated with albuminuria, but not with plasma oncotic pressure or serum albumin concentration, and is abolished if protein intake is decreased, which explains why low-protein diets decrease proteinuria, but do not increase proteinuria. albumin concentration in plasma.
Hypoalbuminemia (albumin less than 3 g / dl) appears when proteinuria and renal catabolism of albumin exceed the hepatic synthesis capacity. The degree of hypoalbuminemia is correlated with the magnitude of proteinuria, although not constantly, since other factors such as age, nutritional status and the type of kidney injury also influence, which justifies that there are patients with very high proteinuria without hypoalbuminemia. This finding is characteristic of some glomerular lesions that present with hyperfiltration, such as obesity nephropathy, reflux nephropathy or secondary to reduced renal mass.
Edema
The appearance of edema is the most visible clinical sign and is usually the reason for consultation, it is a soft, pitting edema that accumulates in declining areas (feet, sacrum) and in regions with small tissue pressure, such as in the periorbital region . If hypoalbuminemia is severe, ascites and pleural effusion may occur. Pulmonary edema does not occur unless there is kidney or heart failure.
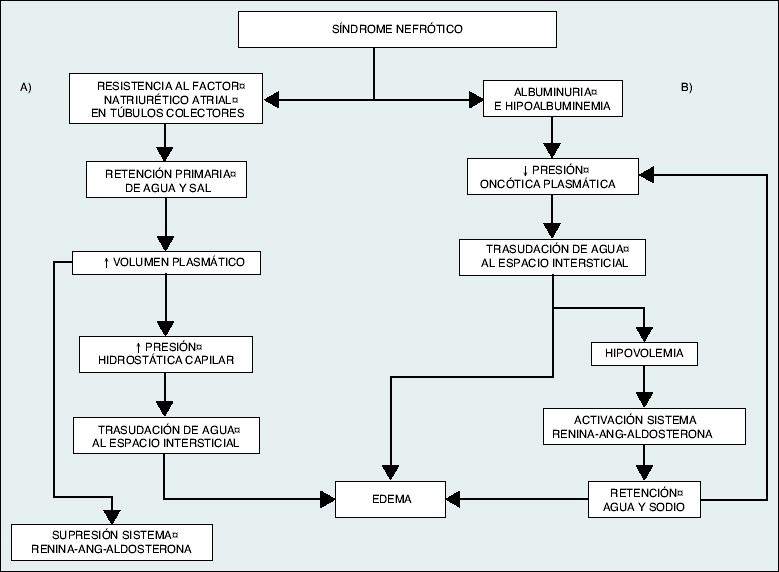
The classical theory is that of hypovolemia, where the renal retention of water and sodium is a consequence of the decrease in plasma oncotic pressure resulting from hypoalbuminemia. This would favor the extravasation of fluid from the intravascular to the interstitial compartment, causing edema. The homeostatic response to hypovolemia would activate the renin-angiotensin-aldosterone system, increase sympathetic tone and the release of antidiuretic hormone. Gradually, the plasma volume would normalize at the expense of an increase in extracellular space and a significant increase in edema.
The alternative theory that would explain the origin of edema, with the theory of volume expansion or hypervolemia. According to this, there would be intrinsic damage to the nephron that would lead to a primary increase in tubular sodium reabsorption, regardless of the hemodynamic situation, inducing a volume expansion that would favor the appearance of edema. The theory of hypervolemia is today the most accepted. In the case of very severe hypoalbuminemia, especially in children, the situation of effective hypovolemia would occur and the mechanisms proposed by the classical or hypovolemic theory would be more important.
|
Pathogenesis of edema formation in nephrotic syndrome. A) Theory of hypervolemia or volume expansion, according to which edema would be the result of the retention of water and sodium by the diseased kidney itself, which would produce an expansion of plasma volume and an increase in capillary hydrostatic pressure that, together with hypoalbuminemia, would favor the transudation of fluid to the interstitial space. B) Classical or hypovolemic theory, according to which the decrease in oncotic pressure secondary to hypoalbuminemia would favor a hypovolemic situation and the retention of water and sodium through the activation of the renin-angiotensin-aldosterone system. This situation is only of clinical significance in circumstances of severe hypoalbuminemia (albumin <2 g / dl). |
Dyslipemia
It is the result of an increase in the hepatic synthesis of lipids and apolipoproteins and, above all, of a decrease in the clearance of chylomicrons, very low density lipoproteins (VLDL), low density (LDL) and intermediate density (IDL) .
Hypercholesterolemia is the most constant lipid abnormality and its severity is inversely correlated with the magnitude of hypoalbuminemia and with the decrease in plasma oncotic pressure. Hypertriglyceridemia, less common, only appears when serum albumin falls below 1–2 g / dl.
In a very characteristic way, there is lipiduria, with fatty casts being observed in the urine sediment. It is advisable to adopt the necessary therapeutic measures to normalize hyperlipidaemia in all patients with nephrotic syndrome, such as lipid-lowering drugs for
Reduce cardiovascular morbidity and mortality or the progression of kidney disease with the correction of lipid abnormalities.
Thrombosis
Renal vein thrombosis and, in general, thromboembolic phenomena constitute one of the most serious complications of nephrotic syndrome. Hypoalbuminemia (serum albumin less than 2.5 g / dl), proteinuria greater than 10 g / 24 h, antithrombin III values less than 75% of normal, and hypovolemia have been associated with an elevated risk of thromboembolic complications.
Infections
There are several factors that condition a high susceptibility to infections in nephrotic patients, including IgG deficiency (due to decreased synthesis and increased leakage losses and renal catabolism), inadequate opsonization due to a decrease in complement factor B, as well as disorders in cellular immunity favored by vitamin D deficiency, malnutrition, and transferrin and zinc deficiencies, both essential for proper lymphocyte function.
Arterial hypertension
Common finding in nephrotic syndrome. The predisposing factors for hypertension are not clearly understood, and may be related to sodium and water retention, or urinary loss of vasodilator (hypotensive) substances.
Endocrine disorders
There is a relative deficit of EPO (Erythropoietin), however, the impact on erythropoiesis is nil, except in isolated cases. Alterations in vitamin D metabolism, decreased thyroxine (T4) and total triiodothyronine (T3) concentrations can also be found, but patients are clinically euthyroid.
Other complications
In long-term nephrotic syndromes, other complications can occur. Thus, persistent proteinuria and increased renal catabolism of filtered proteins trigger a negative nitrogen balance and protein malnutrition. Proximal tubular abnormalities, such as glycosuria, hyperphosphaturia, and Fanconi's syndrome have also been described.
 Glomerular diseases
Glomerular diseases
Initially, we will explain what are in general, the classification of glomerular diseases by time of evolution and then by etiology. We will refer in this chapter, especially to those with Nephrotic Syndrome and then those with predominantly Nephrotic Syndrome. A first clinical approach allows glomerulonephritis to be classified according to its evolution over time. Acute glomerulonephritis evolves in days, with a beginning, and often an end, well defined in time. Rapidly progressive glomerulonephritis is characterized by a rapid deterioration of renal function over weeks or months, with no spontaneous tendency to improvement, with a common histological substrate, which is extracapillary proliferation in the form of epithelial crescents. Chronic glomerulonephritis is characterized by its insidious course and variable evolution over the years. Another approach is etiological, since in many glomerulonephritis a causal factor can be identified. Two large groups are thus distinguished: primary and secondary glomerulonephritis. However, this etiological classification has many limitations, since it does not allow us to distinguish between the different primary glomerulonephritis, whose origin is unknown and, in addition, the same cause can produce several patterns of glomerular disease of different clinical course and prognosis. In the following diagram we will see normal glomerular cellularity and histology, in order to understand the different variants of glomerulopathies that can be found. Another approach is etiological, since in many glomerulonephritis a causal factor can be identified. Two large groups are thus distinguished: primary and secondary glomerulonephritis. However, this etiological classification has many limitations, since it does not allow us to distinguish between the different primary glomerulonephritis, whose origin is unknown and, in addition, the same cause can produce several patterns of glomerular disease of different clinical course and prognosis. In the following diagram we will see normal glomerular cellularity and histology, in order to understand the different variants of glomerulopathies that can be found. Another approach is etiological, since in many glomerulonephritis a causal factor can be identified. Two large groups are thus distinguished: primary and secondary glomerulonephritis. However, this etiological classification has many limitations, since it does not allow us to distinguish between the different primary glomerulonephritis, whose origin is unknown and, in addition, the same cause can produce several patterns of glomerular disease of different clinical course and prognosis. In the following diagram we will see normal glomerular cellularity and histology, in order to understand the different variants of glomerulopathies that can be found.
|
Histological classification of primary glomerulonephritis
|
|
Etiological, histological and clinical classification of glomerulonephritis (GN)
GN associated with systemic diseases
|
Causes of nephrotic syndrome
Any glomerular disease, primary or secondary, can produce nephrotic syndrome at some point in its evolution. The most common causes are listed in Table 1. The diseases that cause nephrotic syndrome vary with age. Thus, the majority of nephrotic syndromes in children are due to minimal change nephropathy. In adults, the most common cause is a secondary form, diabetic nephropathy. The prevalence of primary glomerulonephritis in adults with nephrotic syndrome is variable, depending on the geographic region and the population studied. The most common are membranous nephropathy, sclerosing and focal glomerulonephritis (increasing and the leading cause in blacks), and minimal change nephropathy.
| Etiology of nephrotic syndrome | ||
| Primary glomerulonephritis | Children (%) | Adults (%) |
| Minimal change nephropathy | 52.2 | 14.8 |
| Sclerosing and focal glomerulonephritis | 33.3 | 15.1 |
| Membranous glomerulonephritis | 5.8 | 22.2 |
| Mesangiocapillary glomerulonephritis | 4.3 | 7 |
| IgA nephropathy | - | 4.9 |
| Other primary glomerular lesions | - | 10.3 |
|
Secondary glomerular diseases
|
|
Clinical and laboratory evaluation of nephrotic syndrome
|
Glomerular nephropathies
Lipoid nephrosis or minimal changes:
It is a typical childhood disease (80%), although it can occur at any age. Clinically, it is characterized by alterations of pure nephrotic syndrome, with selective proteinuria. Although it is usually idiopathic, it can, exceptionally, be associated with other diseases. Little functional alteration, except for its presentation in advanced ages, where oliguric renal failure may appear, determined by hypovolemia. Histologically, it is characterized by the absence of relevant histological lesions by light microscopy and the presence of podocyte fusion by electron microscopy. In immunofluorescence, although it is generally negative, some mesangial deposit of IgM or complement can be seen, accompanied or not by minimal mesangial proliferation.
Focal and segmental glomerulosclerosis:
It can occur in adults and children, it can be primary and secondary. It is characterized by the presence of sclerosing lesions in some glomeruli (focal) and only in some capillary loops (segmental) and immune deposits of IgG and C3. Clinically they present nephrotic syndrome with non-selective proteinuria, microhematuria, leukocyturia, arterial hypertension, and variable degrees of renal function impairment.
Glomerulopatía Membranosa:
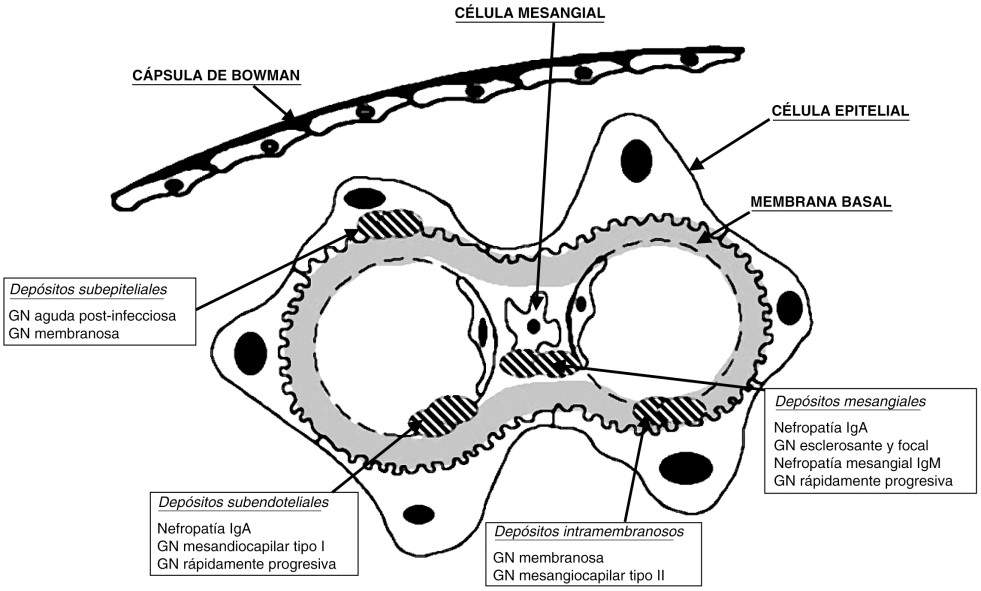
Clinically, it presents intense proteinuria and is the most common cause of nephrotic syndrome in adults. It can be primary and secondary, in secondary forms, it can be associated with Hepatitis B, C viruses and neoplasms. From a morphological point of view, it is characterized by the existence of subepithelial deposits along the basement membrane in the absence of cell proliferation, which gives the histological characteristic of diffuse thickening of the basement membrane. Said deposit is gradual and gives the morphological classification in 4 stages. Between 20-30% of this glomerulopathy is self-limiting. As the injury progresses, the glomeruli become sclerosed and interstitial fibrosis and tubular atrophy occur, leading to CRF in years.
Glomerulopatía Membranoproliferativa:
Also known as mesangiocapillary, it is a chronic glomerular disease, it can be primary and secondary, with a variable clinical course. In the secondary forms, infectious, hereditary or multisystemic diseases are found as factors (SLE, Hepatitis, neoplasms, cryoglobulinemia, etc.). The predominant feature is mesangial proliferation, together with thickening of the glomerular capillary wall and hypocomplementemia (characteristic of a decrease in C3). There are 3 types according to histological classification: Type I, classic form, with subendothelial deposits; type II, with dense intramembranous deposits; Type III, subendothelial and subepithelial deposits. The clinical presentation does not differ in the subtypes, nephrotic syndrome predominates (50%) associated with hematuria and / or asymptomatic urinary abnormalities.
Diffuse Mesangial Proliferative Glomerulopathy:
Uncommon, histologically characterized by non-specific mesangial lesions (diffuse increase in mesangial cellularity and glomerular sclerosis lesions) and clinically presents with nephrotic syndrome with microhematuria. On immunofluorescence, deposits of IgM, IgA, and C3 can be found.
Rapidly Evolving Glomerulonephritis:
It is defined clinically by rapid deterioration of kidney function in weeks or months, with little tendency to spontaneous recovery. The histological characteristic is the extracapillary proliferation in the shape of crescents of more than 50%, its incidence is variable and can occur at any age. There are primary forms (idiopathic, with 3 types) and secondary to autoimmune and infectious diseases.