
 English
English  Português
Português  Español
Español Juan C. Linares Casas
Ischemic or coronary heart disease is a heterogeneous disease that encompasses a wide spectrum of clinical manifestations, ranging from asymptomatic disease -silent ischemia- and stable angina, on the one hand, to unstable angina, acute myocardial infarction, ischemic cardiomyopathy and sudden death, on the other. It is estimated that 30 to 40% of acute coronary events occur in people without a clinical history of ischemic pathology; therefore, the prevention of acute attacks constitutes a real challenge in cardiovascular medicine. A deeper knowledge of its pathophysiological bases can lead us to form more effective prevention strategies.
Pathophysiology.
The usual anatomical substrate - not exclusive - of coronary obstruction is the reduction of the vascular lumen by atheromatous plaques. These plaques are located in the epicardial arteries and will progressively generate a decrease in oxygen supply below a critical level of demand in a given area of the left ventricle.
1.- Decrease in the supply of O2: this supply is controlled by hydraulic and anatomical factors that regulate coronary flow:
a) The hydraulic factors are: coronary perfusion pressure, linked in turn to aortic pressure; left ventricular end-diastolic pressure, and coronary flow time in diastole. As is known, systolic flow to the subepicardial myocardium reaches 30% of the total, but the subendocardium does not receive flow in systole. A restriction to coronary flow in diastole thus produces a marked hypoperfusion of the subendocardium.
b) The anatomical factors are given by the coronary vascular bed and the presence of obstructive atherosclerosis in the large epicardial vessels. The resistance to flow caused by atheromatous stenoses when they exceed a 50% reduction in vascular diameter generates a transstenotic gradient with a drop in the coronary reserve, which will be greater as the reduction in diameter increases.
It should be added here that a rapid acceleration of obstructive lesions is due to the installation of a thrombosis on the atherosclerosis plaque, which can lead to occlusion or subocclusion of the affected artery. Many authors call this condition atherothrombosis, to emphasize the transcendent role of this pathogenic association in acute coronary conditions.
2.- Increased O2 demand: oxygen demand is determined by heart rate, duration of systole, myocardial contractility and left ventricular wall tension.
Any increase in any of these factors will increase oxygen requirements, leading to an increase in the trans-stenotic gradient and a further fall in coronary reserve. This is the case of aortic stenosis, arterial hypertension and tachyarrhythmias.
However, acute and chronic coronary syndromes are active processes and not limited to a static environment of fixed atheromatous plaques. Its causative agents are actually abnormalities of a hostile molecular and biological environment for the vascular endothelium, an environment that is stimulated by a system of signals that alter its integrity, triggering various inflammatory reactions. The atheroma plaque is today conceived as the seat of biochemical reactions between the single cell endothelium, the underlying smooth muscle cell and the blood elements that, at another time, circulate normally. Thanks to the understanding of these cellular interactions and the magnitude of their damage, we can now differentiate stable atheromatous plaque from unstable or vulnerable one.
Atherosclerosis is, in synthesis, the consequence of an interaction between different aggressive agents (dyslipidemia, tobacco, hypertension, diabetes) and very complex defensive responses. As a result of this "fight" the arterial wall undergoes progressive structural and functional alterations.
The normal and pathological endothelium.
Despite its microscopic dimensions, the vascular endothelium is an extremely important endocrine organ, whose integrity is a fundamental requirement to preserve the structure and function of the vascular wall, as well as the regulation of thrombotic phenomena. In a 70 kg man, it has an area equivalent to 6 tennis courts, a total weight of more than two kilos - more than the liver - and a total number of cells in that monolayer of a trillion units. These cells receive signals and emit signals. They "sensan" changes in flow, pressure, inflammatory signals, or levels of hormones or functioning molecules, and they respond by synthesizing, releasing, and activating substances that regulate four primary functions:
Vascular tone
The growth and proliferation of smooth muscle cells
Thrombosis and homeostasis
Inflammatory and immune response
As an important metabolic and endocrine organ, it acts by regulating these functions in a dual way: it secretes relaxing factors and vascular constrictors, thrombogenic and antithrombotic, fibrinolytic and antifibrinolytic, inhibitors and stimulants of cell growth, etc.
But we must emphasize that, under normal conditions, it works in an inhibitory way, facilitating circulation: it secretes vasodilator factors, inhibits vascular smooth muscle contraction, thrombosis, platelet aggregation, cell growth, and leukocyte adhesion. But under pathological conditions, when the endothelium is injured by being attacked by different noxas, it is “activated” and its function will be the opposite: its surface will stimulate coagulation and cell adhesion, while producing vasoconstrictor substances and stimulants of cell growth that they will thicken your wall.
Vasorelaxant substances include nitric oxide and prostacyclin, which are both anti-inflammatory, growth inhibitor and, in the case of prostacyclin, antithrombotic. Transforming growth factor beta, thrombospondin, and heparan sulfate also inhibit cell growth. Antithrombotics include urokinase, heparin proteoglycans, tissue plasminogen activator, and thrombomodulin.
This endothelial dysfunction characterizes the initial stage of atherosclerosis, when vascular structural changes have not yet occurred. The balance between the relaxing and constrictive endothelial factors has been unbalanced, with a predominance of the latter: endothelin, angiotensin II, platelet-derived growth factor (PDGF), as well as pro-thrombotic substances (tissue factor, platelet activating factor, Von Willebrand factor, tissue plasminogen activator inhibitor), vascular growth factors, and pro-inflammatory cytokines.
Deficient or altered nitric oxide (NO) production has been shown to be largely responsible for endothelial dysfunction, in response to certain pathological stimuli. These biochemical and biophysical stimuli are capable of unbalancing endothelial homeostasis, leading to an increase in the permeability of atherogenic lipoproteins, increased adhesion of monocytes, stimulation in the production of cytokines and advanced end products of biosynthetic reactions, as well as the alteration in the hemodynamic and biomechanical forces that lead to greater endothelial damage. All of this results in thrombosis, inflammation, abnormal vascular reactivity, intimal hyperplasia, and fibrosis.
The fatty streak will be the first histopathological manifestation of atherosclerosis disease, which will progress until the fibrous plaque is formed, which consists of a lipid nucleus covered by a fibrous layer that results from the synthesis of collagen, elastin and proteoglycans by muscle cells. smooth and macrophages that have migrated to the arterial intima.
The concept of Angina Pectoris
The term "angina pectoris" or "angor pectoris" was introduced by Heberden in 1768 to indicate a very characteristic "disorder of the chest" accompanied by "feeling of strangulation and anxiety". By using the word “angina” (strangulation), Heberden described it as a type of chest pain of a very special modality that is accompanied by psychic phenomena, the most notable of which consisted of the fear of imminent death (angor animi ).
Angina pectoris is a syndrome in itself, not a disease. It always indicates the same basic pathophysiological disorder: myocardial ischemia, whatever the cause that generates it.
Although it is almost impossible to find a clear definition of angina pectoris in the publications, it can be established that it is a clinical syndrome characterized by crisis of pain, tightness or discomfort of ischemic origin, generally located behind the sternum, but located at sometimes in the jaw, shoulders, back or arms, aggravated or triggered by effort or emotions, and which is rapidly relieved when the activity or causal disturbance ceases, or by the administration of nitroglycerin.
Some elements described above may not be typical, but the oppressive characteristic of the pain or discomfort is the basis for their recognition. The location, radiation or duration may vary, and may not even be related to efforts, but the oppressive, constrictive sensation of heaviness constitutes the most precise character of the angina.
Pain characteristics.
The location is usually retrosternal, although the initial site is sometimes broader and includes most of the precordial region. Sometimes it is not located in the thorax: it can affect the jaw, the left arm, the epigastrium, the shoulders, the scapular region and other atypical areas. In these cases, its paroxysmal appearance after exertion, its oppressive nature, and its rapid cessation with rest suggest it.
Although it may be located behind the breastbone, the pain tends to radiate to the neck, lower jaw, and upper extremities. In its most classic form it radiates towards the scapula and upper part of the left arm. Sometimes the distribution of the ulnar nerve along the antero-medial aspect of the hands and fingers clearly follows.
As for its character, what has been said is reiterated: it is a painting that is classically constrictive or oppressive. It has also been described as choking or burning. There are times when patients do not report pain but only tightness or the sensation of a weight on the chest. It can be light or intense, but its quality is characteristic. In many cases, the pain so mimics the strain behind the breastbone that the patient tries to vomit for relief.
It usually forces the patient to keep as still as possible and, if he is marching, he must stop. It is usually accompanied, as already mentioned, by a feeling of imminent death, which is not related to its intensity. As crises occur over time, patients stop feeling this anguish, due to the greater knowledge or experience they have acquired in the face of the disease.
Angina episodes usually begin gradually, reach their maximum intensity soon, and also gradually disappear in a few minutes, usually two to ten. They are relieved quickly with the cessation of the activity that caused it, rest or the administration of sublingual nitroglycerin. If the pain lasts thirty minutes or more, the installation of an acute coronary accident (unstable angina or myocardial infarction) should be suspected.
Triggers.
Exertion is the most important and frequent triggering cause of angina pectoris, as it increases the oxygen demand. The most common physical exercise is walking or climbing stairs. The severity of the condition is related to the effort required to generate pain.
Exposure to cold and digestion increase the possibility of angina after exertion that is not usually followed by pain. Likewise, emotional states can trigger anginal crises through complex adrenergic mechanisms.
But it is important to note that in many patients the episodes of pain occur without apparent cause and at rest, as a result of a sudden drop in the supply of oxygen to the myocardium, as a consequence of a rupture of the atheromatous plaque with added thrombosis (coronary accident acute), or as an expression of a dynamic coronary obstruction (spasm). We will return to these points later.
The frequency with which angina presents in an atypical form and the large number of diseases that present with chest pain require a careful and often difficult differential diagnosis. The stabbing, fleeting and localized pains in the precordium are usually of psychic origin. The pain of aortic dissection, pneumothorax and, in general, of processes that occur with rupture or laceration of the tissues, begins abruptly and its intensity is maximum from the beginning.
The circumstances and symptoms that accompany the pain are very useful in the differential diagnosis. The relationship with food intake or improvement with alkalis will lead to a digestive disorder; changes in intensity with movement of the upper limbs or neck and with breathing or body position suggest cervical radiculopathy or pericarditis, respectively. By contrast, anginal, oppressive, and distressing pain is often accompanied by a sensation of gravity and, occasionally, sweating and palpitations. Dyspnea during pain is rare, but its appearance indicates a serious coronary disease and is a poor prognostic sign. The presence of risk factors - hyperlipidemia, diabetes, hypertension, smoking,
Clinical classification of angina pectoris
The circumstances in which anginal pain appears broadly indicate the mechanism that causes it and, in turn, knowledge of it allows guiding treatment. Based on these criteria, different classifications have been proposed. We will consider here: a) stable angina pectoris; b) unstable angina pectoris, which is part of the acute coronary syndromes, and c) vasospastic or Prinzmetal's angina.
Stable angina pectoris.
The diagnosis of stable angina pectoris is made by the relationship between coronary pain and exercise. It is defined as one that did not change its characteristics in the last 3 months, with or without a history of myocardial infarction. It constitutes 60% of the diagnosed cases.
In general, the level of effort required to cause angina, or angina threshold, is constant over long periods of time, so the patient usually knows in advance what activities in their daily life will cause it. When a patient with stable angina changes the characteristics of his pain - more frequent appearance, less intense provocative efforts, etc. - he will have entered the group of unstable angina.
According to its severity and the functional limitation it imposes on the patient, exertional angina is divided into four grades following the Canadian Cardiovascular Society classification:
Grade I : regular physical activity does not produce angina, which appears with significant, rapid and / or prolonged efforts.
Grade II : mild limitation of physical activity; the pain appears when walking at a normal pace two or three blocks or going up more than one floor quickly, walking on slopes, in the postprandial period, or in cold climates or against the wind; in emotional states or when starting morning activity.
Grade III : significant limitation of usual activity: pain occurs when climbing a floor or walking a block with a normal gait.
Grade IV : inability to carry out any physical activity without the appearance of angina; angina may occur at rest.
Almost 80% of patients with stable angina belong to classes I and II. Grade III-IV patients have been shown to have higher mortality than those in grades I and II.
Physical examination is often normal, especially after the seizure has passed. During anginal access, pallor, diaphoresis, tachycardia may appear, as well as the auscultation of a fourth noise. The presence of hypotension and heart failure during attacks are signs of severity.
The electrocardiogram is normal in 50% of cases, in the absence of pain. The remainder may show signs of an old infarction, left ventricular hypertrophy, ST segment depression, or ischemic changes in the T wave.
The ergometric test constitutes a very important diagnostic test in these patients. It is considered positive if during the same, or immediately after, angina appears or if the ST segment descends at least 1 mm adopting a horizontal shape. If the pain appears early (less than 6 minutes) or if the ST depression exceeds 2 mm or the blood pressure drops during exertion, we will be dealing with a high-risk patient who should be referred for a coronary angiographic study with a view to revascularization.
Radioisotopic imaging studies and stress echocardiography can identify the extent, severity, and location of the ischemia. Coronary angiography and multislice tomography will inform us about the status of ventricular function and the extent of coronary lesions. It is important to highlight that survival is related to the degree of coronary atherosclerosis: the greater the number of obstructed coronary arteries and the greater the contractile deterioration of the left ventricle, the worse the prognosis. In this sense, coronary angiography findings are the best predictive factors.
Regarding the coronary morphology in stable angina, it should be emphasized that the images of the atheromatous plaques are those of an uncomplicated atheroma, that is, with a smooth, yellowish-white surface, without ulcerations, intimal hemorrhages or adherent thrombi. In this indemnity of the fibrous covering of the plaque lies the difference with the lesions of the unstable type.
Unstable angina pectoris.
Some types of angina pectoris are considered unstable forms of coronary heart disease, and the therapeutic approach differs significantly from that of stable angina, since their presence indicates a situation of unpredictable evolution. Under the term unstable angina we can include the following types: a) recent-onset angina (duration of symptoms less than one month); b) progressive angina: it is one where the painful crises become more frequent or of longer duration, are rebellious to nitroglycerin or appear with less and less intense efforts; c) the third subgroup is constituted by angina at rest.
Unstable angina is part of the so-called group of acute coronary syndromes, for which it will be analyzed later.
Prinzmetal's angina.
Prinzmetal's angina or variant angina is a particular clinical manifestation of ischemic heart disease characterized by painful attacks at rest and elevation of the ST segment during them (sometimes in the form of a monophasic wave).
This condition responds to an increase in coronary vasomotor tone or a spasm in the vessel, which can settle on a healthy or atheromatous coronary artery, and which will cause ischemia through a decrease in oxygen supply.
The electrocardiogram is usually normal outside of anginal seizures, especially in the absence of coronary atherosclerosis.
Patients with coronary spasm present a very marked response to the administration of ergonovine, which reproduces the clinical and electrocardiographic picture of variant angina. It is, however, a potentially dangerous test that should be avoided if the state of the coronary tree is not known.
ACUTE CORONARY SYNDROMES
Acute coronary syndromes (ACS) are a frequent cause of consultation in daily medical practice. Its importance lies in the high morbidity and mortality and the high healthcare cost it generates, the latter being linked to the use of medication and the performance of paraclinical tests and the performance of invasive diagnostic and therapeutic procedures.
Definition and classification
SCA is an operational term that has been developed to refer to a constellation of symptoms consistent with acute myocardial ischemia. This encompasses a varied spectrum of clinical entities ranging from unstable angina (AI) to acute myocardial infarction (AMI) in all its variants.
ACS are classified according to the ST segment abnormalities of the initial electrocardiogram (ECG) in:
- ACS with ST segment elevation; Y
- ACS without ST segment elevation.
This is justified based on the fact that the first group of patients will be considered as having an acute infarction and the strategy of greatest therapeutic importance is emergency myocardial reperfusion through thrombolysis or angioplasty.
In the second group, patients with unstable angina and AMI without ST segment elevation are presented, differing from each other by the absence or presence, respectively, of biological and enzymatic markers in the blood; likewise, the initial therapeutic management is the same for both entities.
Pathophysiology of acute coronary syndromes
ACS share a common pathophysiological origin (Fig. 1): the accident of the atherosclerosis plaque, with erosion or rupture of its fibrous covering and, as a consequence of various types of injury, hemodynamic forces and probably inflammation, exposes a highly thrombogenic substrate that When interacting with the blood, it generates two outstanding events a) platelet activation and aggregation and b) the generation of thrombin. Both events interact and give rise to a thrombus of greater or lesser magnitude.
If the thrombus is occlusive, it will generate the total interruption of the coronary flow in the culprit vessel and usually the consequence is transmural myocardial infarction or Q-infarction (with elevation of the ST segment), whereas if the added thrombosis is not occlusive, the picture is usually The accompanying clinical picture is non-ST elevation AIS, that is, unstable angina or non-Q infarction.
The vulnerable or high-risk plaque.
Despite the fact that atheromatous lesions have the same pathophysiology, the plaques are very heterogeneous and those "high risk" have their own characteristics. The knowledge of atherosclerosis disease advanced from the old idea that maintained that the acute occlusion of an artery was the final consequence of a slowly progressive obstruction, to the current concept that establishes that it is the product of the rupture of a plaque, with subsequent formation of a thrombus that partially or totally occludes the arterial lumen. But only recently has it been postulated that the composition of the plaque is more important as an indicator of rupture risk than the previous degree of obstruction, becoming the determining factor of this disease.
The atherosclerosing plaque prone to rupture, also called "vulnerable" or "high risk" has, from the histological point of view, a large lipid nucleus, usually eccentric and with extracellular lipid deposits, high density of T-lymphocytes and macrophages full of lipids, a reduced number of smooth muscle cells, and all covered by a thin fibrous layer (Fig. 1). Its consistency is that of a toothpaste at room temperature, being even less consistent at body temperature. Therefore, it is not surprising that these plaques are not very stable and prone to rupture, even more so when compared to other fibrous plaques rich in collagen (Fig. 2).
Once the thin fibrous layer is broken or eroded, its lipid interior is exposed to the blood stream that triggers the coagulation cascade, generating a thrombus that occludes or obstructs the lumen, with the clinical manifestations of acute coronary syndromes (Fig. 3 ).
 Figure 1
Figure 1
 Figure. 2: The stable plaque has a relatively thick fibrous layer that protects the lipid core from contact with blood.
Figure. 2: The stable plaque has a relatively thick fibrous layer that protects the lipid core from contact with blood.
 Figure 3: Physiopathological schematization of Acute Ischemic Syndromes.
Figure 3: Physiopathological schematization of Acute Ischemic Syndromes.
Q AMI: type Q or transmural myocardial infarction; AI: unstable angina;
Non-Q MI: non-Q type acute myocardial infarction.
Unstable angina suggests that there has been a relatively small erosion or fissure of a vulnerable plaque that gives rise to transient episodes of thrombotic occlusion with the consequent angina at rest. This thrombus is usually labile, with occlusions that do not exceed 15-20 minutes. In addition, the release of vasoactive substances by platelets will cause secondary vasoconstriction, which may contribute to further reducing coronary flow.
In non-Q infarction, plaque damage is more important and results in more persistent thrombotic occlusions, which can last up to an hour. A quarter of patients with non-Q infarction have coronary occlusion for more than one hour, but the ischemic myocardial territory is usually irrigated by collaterals.
In type Q or transmural infarction, a major plaque fracture occurs that can result in the formation of a fixed and persistent thrombus for more than one hour with the consequent transmural necrosis of the compromised myocardium. Some cases of sudden coronary death are probably based on a rapidly progressive lesion where the rupture of the plaque and the resulting thrombosis lead to fatal ventricular arrhythmias.
Clinic
Patients with unstable angina and myocardial infarction often complain of anginal pain with characteristics similar to those of stable angina, but are more severe and prolonged. Just as in classic exertional angina the pain lasts between 2 and 10 minutes, approximately, in unstable angina it persists between 10 and 25 minutes, appears with less intense efforts and even at rest.
In myocardial infarction, anginal pain usually lasts more than 30 minutes - sometimes hours - and is often more intense. It is not relieved by nitroglycerin and may be accompanied by arrhythmias or symptoms and signs of heart failure. It does not intensify when sitting or breathing deeply, a useful criterion to differentiate it from the pain of acute pericarditis.
Most heart attack patients, especially those with ST segment elevation, are distressed and agitated, and they try unsuccessfully to relieve their pain by moving in bed, modifying their posture. It is common to find paleness along with sweating and coldness of the extremities. The combination of retrosternal chest pain lasting more than 30 minutes and sweating is a strong argument in favor of an acute myocardial infarction. In addition, an apexian presystolic gallop (R4) is usually heard, due to the forceful contraction of the left atrium due to the increase in left ventricular diastolic pressure.
Blood pressure and heart rate may show hypertension and tachycardia or hypotension and bradycardia, depending on whether there is sympathetic or parasympathetic hyperactivity, respectively, although in ST-elevation infarcts the systolic pressure usually falls by about 10 or 15 mm Hg.
After the first days after the infarction, there is a pericardial rub in 6 to 10% of cases, which is usually intermittent and is heard more clearly at the apex or at the left sternal border; it may persist for several days and is due to localized pericarditis due to the transmural extension of the necrosis to the epicardium.
Fever usually occurs within 24 hours of the infarction, generally of moderate degree and lasting between two and four days. Sometimes there is vomiting, which is sometimes due to the medication used.
In about half of the cases a trigger factor is detected prior to the heart attack, in general an intense and unusual physical exercise, an emotional stress or a medical or surgical disease. It can occur at any time of the day or night, but its frequency reaches its highest value in the first hours of awakening. This circadian peak is due to the combination of increased sympathetic tone and a greater tendency to thrombosis - greater platelet aggregability - which occurs between 6 a.m. and 12 noon.
In the case of left ventricular dysfunction, cardiogenic shock and ischemic mitral regurgitation, the signs of such complications will be found.
Before going any further, we must underline a point that should not be forgotten: around 25% of heart attacks are not recognized clinically; half of them are asymptomatic and the diagnosis is made retrospectively by analyzing an electrocardiogram. In the rest, the pain is atypical or not present. Some authors give even higher percentages of "silent" heart attacks.
Electrocardiogram.
The electrocardiogram (ECG) is of paramount importance and allows ACS to be classified into two large groups: with and without ST segment elevation. The former will usually develop a Q wave myocardial infarction (transmural infarction), while the latter will most likely have unstable angina or a non-Q wave infarction. The difference between the latter two conditions will be given by the presence or absence of biochemical markers of necrosis in blood tests, as will be seen later.
As long as a sufficient thickness of the left ventricular wall is affected, and more specifically in transmural infarcts, changes in the ECG occur due to: a) ischemia; b) more advanced myocardial damage or an area called “injury”, and c) necrosis or infarction itself. These alterations develop dynamically and in an evolutionary way, successively, conditioning the following modifications:
Initially, in the first minutes of coronary occlusion, ischemia modifies ventricular repolarization and produces inversion of the T waves, which will appear negative, pointed and deep in the leads that face the ischemic area. Therefore, in anterior wall infarcts, T are probably negative in precordial leads; in lateral infarcts, they will be in D1 and aVL; and in posteroinferior infarcts the T will be inverted in D2, D3 and aVF.
As the ischemia and consecutive myocardial damage are accentuated, the so-called "injury currents" or "injury" appear in the leads that look at the injury, and which are translated on the ECG by an elevation of the ST segment, an elevation that reaches its maximum expression on the 1st or 2nd day of the event. This ST elevation can even become monophasic wave type (Fig. 4).
The area of myocardial necrosis does not generate action potentials. If the infarct covers the entire thickness of the ventricular wall (transmural infarction), it can be considered as an open window in the wall. Therefore, the negative potential within the ventricular chamber is transmitted through the necrotic tissue to the leads that face the infarcted area. Instead of the normal positive wave, the beginning of the ventricular QRS complex will be directed downwards, that is, there will be deep and abnormal Q waves, and only negative electrocardiographic complexes of the QS type. These Q waves are wider and deeper than the Q waves normally seen in some leads. They appear several hours after the beginning of the picture, succeeding the changes in the ST segment. Its late appearance obliges, therefore,
The wave of injury or positive ST segment elevation is therefore essential for the diagnosis of transmural infarction and its extension is proportional to its size or extension. This injured tissue appears from the first hours after the occlusion and persists, usually in gradual decline, for 3 to 4 weeks, evolving towards total necrosis (deep Q waves) or, at least in part, towards simple ischemia, indicating in the latter case a better evolution. As the ST elevation improves, there will then be an increase in ischemia in the subepicardium (negative T waves).
In the case of non-ST segment elevation acute coronary syndromes, that is, unstable angina and non-Q infarction.
In unstable angina, the ECG may show no changes, but in most cases transient alterations can be observed: ST segment depression greater than 1 mm or greater than 0.5 mm, inversion of the T waves, or even blockade of the ST segment. left branch of advanced degree and arrhythmias.
In non-Q infarction, it happens that the necrotic tissue does not occupy the entire thickness of the ventricular wall, and therefore the activation wave can reach the healthy transmural area through its usual path. The electrical “window” of Q infarcts does not take place here. We can observe a decrease in the voltage of the R waves in the leads that look at the infarct zone, ST segment deviation, inverted T waves (Fig. 5) and blockade of the branch, as in unstable angina.
The difference between the different acute coronary conditions without ST segment elevation should not be found in the ECG but in the biochemical markers of necrosis.
 Figure 4: ECG of an acute infarction with elevation of the ST segment, evident in leads II, III and aVF
Figure 4: ECG of an acute infarction with elevation of the ST segment, evident in leads II, III and aVF
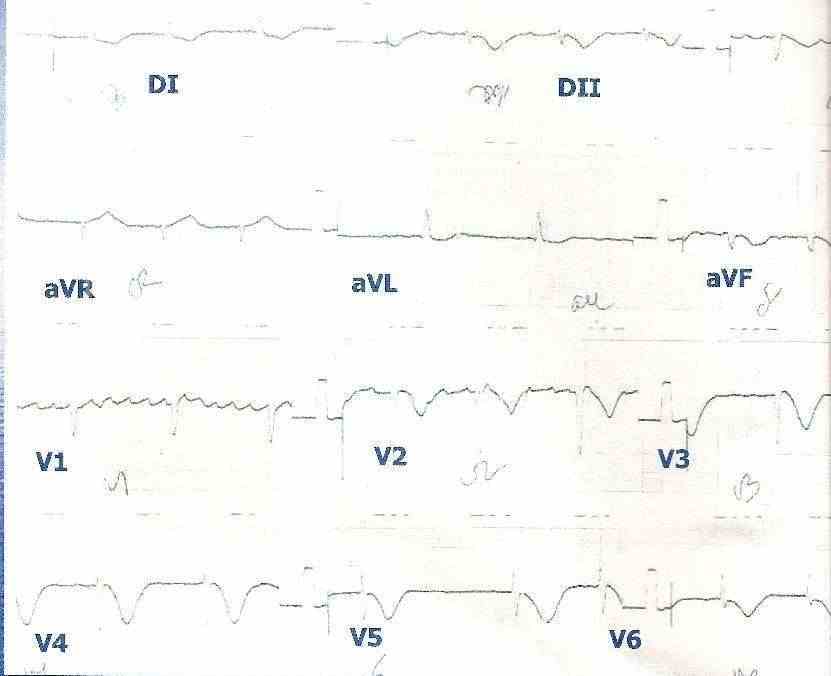 Figure 5: Non-ST segment elevation acute coronary syndrome in a patient with chronic atrial fibrillation. The inversion of the T waves can be observed in leads V2, V3, V4, V5 and V6.
Figure 5: Non-ST segment elevation acute coronary syndrome in a patient with chronic atrial fibrillation. The inversion of the T waves can be observed in leads V2, V3, V4, V5 and V6.
Biochemical markers of myocardial necrosis
Myocardial infarction causes various humoral changes, such as leukocytosis and increased erythrocyte sedimentation rate. However, from a diagnostic point of view, only the appearance in the blood of different proteins released into the circulatory stream, released by damaged myosites, is important: myoglobin, cardiac troponins T and I, creatine phosphokinase (CPK), lactic dehydrogenase (LDH) and others. . The diagnosis of AMI is established when the hematic levels of specific and sensitive biomarkers, such as myoglobin, troponins, and the MB fraction of CPK, are increased in the clinical setting of acute ischemia. These markers reflect myocardial damage but do not indicate its mechanism. Thus,
Two markers are generally used. The most recommended is to use a combination of a rapidly rising marker, such as myoglobin, and another that takes longer to rise but is more specific, such as troponins, in order to detect the presence of infarction in both patients who present early as late.
Myoglobin is detectable in the blood two to three hours after onset. Its concentration rises rapidly, reaches a maximum level between 6 and 12 hours after the onset of symptoms and falls to normal values in the next 24 hours, eliminated by the kidneys.
Cardiac troponins are considered the most commonly used markers of cardiac specificity in the diagnosis of myocardial injury, particularly troponin I and troponin T. These proteins are associated with specific amino acid sequences encoded by genes other than those that encode skeletal muscle isoforms. Thus, TnI has complete specificity for the myocardium, whereas TnT occurs in small amounts in skeletal muscle during human fetal development and is re-expressed in pathologies that are associated with muscle regeneration (for example, muscular dystrophy of Duchenne). Troponins appear in the serum relatively early after the onset of the infarction (4 to 10 hours), reach their peak in 48 hours, and remain abnormal for 4 to 10 days.
If troponin determination is not available, the best alternative is CPK-MB: it is less specific than cardiac troponin, but its clinical specificity for irreversible injury is more robust. As with troponins, an increased CPK-MB (for example, for the diagnosis of AMI) is one that exceeds 99% of the values of a control group.
Measurements of total CPK are not usually recommended for the routine diagnosis of infarction, due to the wide tissue distribution of this enzyme. However, its long history means that many doctors still use it, although in these cases it is desirable to combine it with a more sensitive biomarker. The prior use of intramuscular injections should always be ruled out.
Transaminases and lactic dehydrogenase and their isoenzymes should not be used for the diagnosis of acute infarction, since their rise can occur in other clinical circumstances: pulmonary embolism, acidosis, liver and muscle injuries, as well as due to the administration of some medications such as statins. They will only be used when it is impossible to have more specific markers.
Thus, in an acute coronary syndrome without ST-segment elevation, the presence of abnormal biochemical markers will tell us about the existence of unstable angina, while an elevation of them will confirm that we are facing a non-Q infarction.
Echocardiogram
Echocardiography, in the acute stage of coronary events, discovers abnormalities in the motility of the walls in 89% to 100% of patients with transmural infarction, with a decrease in sensitivity between 79% and 86% 6 for non-infarcts. trasmural. Small infarcts may not produce echocardiographic abnormalities. There are technical limitations due to poor acoustic windows and the presence of entities such as conduction blocks and myocarditis.
Forecast
Patients can be classified into 3 risk groups:
a) High risk . Those with angina at rest, recurrent or accelerated and who also present one of the following characteristics: hemodynamic instability (hypotension, heart failure, mitral dysfunction); ST descent> 1 mm transient or persistent; new bundle branch block or ventricular arrhythmias; postinfarction angina (in the first month); after coronary revascularization (in the first month); age> 75 years; markedly elevated troponins (troponin T> 0.1 ng / ml). These types of patients have a greater than 5 percent risk of suffering a myocardial infarction or dying in the first 30 days, so cardiac catheterization is recommended in the next 24 hours with a view to revascularization.
b) Medium risk . Patients with unstable angina plus the absence of high-risk characteristics, T-wave inversion> 2 mm in 2 or more leads, and the presence of some risk modifier (previous coronary artery disease, known peripheral arterial disease or cerebrovascular disease, diabetes mellitus, previous revascularization, troponins> 0.01 ng / ml).
c) Low risk . The rest of the patients without high or medium risk characteristics, and a normal electrocardiogram.
Heart attack complications
Complications of acute myocardial infarction are very common. They can appear at the time of triggering the episode or develop later, but it is in the first days when it occurs most frequently.
These complications are: 1) arrhythmias and atrioventricular conduction disorders; 2) heart failure; 3) cardiogenic soc; 4) acute mitral regurgitation; 5) acute ventricular septal defect; 6) myocardial rupture; 7) pericarditis and post-infarction syndrome; 8) post-infarction angina; 9) right ventricular infarction; 10) pulmonary embolism, and 11) systematic embolism.
1. Arrhythmias . The incidence of arrhythmias during the course of an acute myocardial infarction varies according to the different authors between 70 and 100% of cases. This depends on many factors (age, drug use, acidosis, size of the infarction, etc.) but it is fundamentally given by the different detection methods, since when oscilloscope monitoring is used, its frequency of appearance is lower than when it is used. does with registration in electronic memory or magnetic tape. Following Lown, it is useful to divide arrhythmias into two types: 1) those produced as a result of the ischemic process (electrical failure), and 2) those secondary to a decompensation of the hemodynamic state due to severe myocardial deterioration (mechanical failure).
Ultimately, both are the consequence of an electrophysiological alteration of the myofibrils, but in the latter, the therapy of pump failure must also be addressed.
Arrhythmias will be briefly described on the basis of their autonomous site of origin.
a) Sinus tachycardia. It denotes sympathetic hyperactivity and is found in 30% of the cases; it is more frequent in anterior infarcts than in inferior ones. The most common causes are: pain, anguish, heart failure, pericarditis and hypovolemia. Those that require a precise differential diagnosis are pump failure and hypovolemia, since the treatment is very different in both. A very useful piece of information is the recording of pulmonary capillary pressure with a Swan-Ganz catheter: its measurement will decide whether there is venocapillary hypertension (more than 18 mm Hg) or, on the contrary, hypotension (less than 19 mm Hg.)
b) Sinus bradycardia. In the initial moment of infarction, it occurs in 40% of cases, but in the coronary unit it is observed in only 20%. It is more frequent in lower infarcts than in previous ones, in a ratio of 3: 1, possibly due to parasympathetic hyperactivity.
It is an arrhythmia due to electrical instability, due to sinus node ischemia, vagal reflexes or pericarditis. Sometimes it is associated with ventricular extrasystole due to increased automatism
c) Atrial extrasystole. It occurs in 20% of cases and is due to an increase in automaticity or atrial reentry phenomena, secondary to pump failure, hypoxia, or electrolyte disorders.
d) Atrial fibrillation. It is seen in 10 to 15% of patients and can indicate early left ventricular failure or atrial infarction. It can also respond to extracardiac causes, such as hypokalemia, hyperthyroidism, hypoxia, or pulmonary embolism. As atrial systole contributes to 15% of the ventricular ejection fraction, its loss in an infarcted patient may be essential for the maintenance of an appropriate cardiac output. Likewise, if the ventricular response is high (greater than 130 beats / minute), the hemodynamic state will deteriorate, observing signs of cardiac decompensation (oliguria, basal rales, hypotension) due to a shortening of the diastolic filling period and an increase in myocardial oxygen consumption. which in turn can spread necrosis.
e) Atrial flutter. Present in 1% to 2% of AMI, like fibrillation, it can be due to left ventricular failure or atrial infarction. Depending on the degree of AV block, the heart rate will range from 75 to 300 beats / minute. In inferior infarcts, 3: 1 and 4: 1 blocks are usually observed due to ischemia or injury to the AV node. With these degrees of blocking the typical flapping waves are well evidenced.
f) Paroxysmal atrial tachycardia. It occurs in 5% of infarcted patients. The heart rate ranges between 150 and 300 beats per minute and its pathophysiology is the same as in atrial extrasystole.
g) Rhythm of the AV junction (nodal). Its frequency ranges between 2% and 15% according to different authors. It constitutes an escape rhythm, due to depression of the sinus node or AV block as a consequence of ischemia.
h) Atrioventricular block (AV block). First degree AV block occurs in 9% of cases, second degree in 8% and third degree in 7% of patients admitted to the Coronary Unit with an AMI.
Of the entire atrioventricular conduction system, the AV junction is the sector most sensitive to. anoxia and ischemia. For this reason, in acute inferior infarcts it is common to observe different degrees of AV conduction disorders as a consequence of the compromise of the normal blood supply of the AV node.
First degree AV block (all stimuli cross the AV junction, but do so more slowly), or AV conduction time of more than 0.20 seconds, does not require treatment but does require careful control, as it can evolve to higher degrees lock; the latter is seen more frequently in previous heart attacks.
Second-degree AV blocks (not all stimuli cross the junction) respond to two basic mechanisms. In Mobitz type I (or blocks with Wenckebach periods) a progressive prolongation of the PR segment is observed, which indicates an increasingly affected conduction, until a P wave is totally blocked. This prolongs the time available for the AV node to recover, and the following P wave will present normal AV conduction, which gives rise to a new cycle of progressive PR prolongation.
This type of AV block is more frequent in inferior infarcts and is usually due to ischemia of the AV node. Even without treatment it can disappear in 72 hours. Mobitz I type AV block constitutes 90% of second degree AV blocks.
Mobitz II type second-degree AV block is less common and is seen in less than 10% of all patients with AMI who have a block. It shows the lack of abrupt conduction of a P wave without prior lengthening of the PR segment. In most cases, it is associated with an anterior infarction and is almost always due to injury below the bundle of His, which often progresses to complete blockage. Therefore, this disorder requires, in general, the implantation of a temporary pacemaker in the right ventricle.
In third-degree (complete) AV block, the atria and ventricles beat independently.
It is seen in patients with inferior or anterior infarcts, and it originates in the edema that surrounds the infarcted tissue and compromises the conduction system. In inferior infarcts the lesion is nodal, with gradual development of the block and the appearance of rhythms of the AV junction of 40 to 60 beats per minute. In anterior infarcts, AV block settles abruptly and is a cause of high mortality. It is caused by extensive necrosis of the septum involving the bundle of His (infranodal lesion) and can suddenly end in asystole, pump failure, or ventricular fibrillation. Third-degree AV block is a formal indication for a temporary endocavitary pacemaker and pacing on demand.
i) Intraventricular conduction disorders. They are seen in 10 to 20% of cases. They can be: 1) complete right bundle branch block (RBBB); 2) complete left bundle branch block (LBBB); 3) left anterior hemiblock (HAJ); 4) left posterior hemiblock (HPI); 5) BCRD with HAI, and 6) BCRD with HPI.
Complete bundle branch blocks can progress to third degree AV block in the course of an acute infarction. AMI complicated with RBBB or RBBB have high hospital mortality.
j) Ventricular extrasystole. It is the most common observed arrhythmia in AMI, with a frequency greater than 75% of cases. It is due to an electrical imbalance at the cellular level produced by ischemia (arrhythmia due to electrical instability). As in general there is no hemodynamic decompensation, they are not a bad prognosis, but they should be treated if they appear more than six per minute, if they are multifocal, bigeminate or trigeminated or if they have previously caused serious arrhythmias.
k) Ventricular tachycardia. It occurs in 20% of patients in the first hours of the acute infarction. It is generally defined as a succession of ventricular beats (four or more) with a rate less than 120 beats / minute.
It is more frequent in cases of transmural infarction and in those with severe ventricular failure, and the electrophysiological mechanism responds to the increased automatism of the injured Purkinje fibers (ectopic focus) and reentry phenomena. It is a serious arrhythmia that requires quick treatment decisions.
l) Accelerated idioventricular rhythm. It is defined as a ventricular rhythm with a rate of 60 to 100 beats per minute, and it occurs in 15% of patients with AMI. It is observed on the second or third day of the event, and responds to an escape mechanism, due to a slowing of the base rhythm or to an ectopic focus of ventricular tachycardia. The episodes are usually temporary and have a good prognosis.
m) Ventricular fibrillation. This serious arrhythmia is manifested on the ECG by a succession of slow, polymorphous, and irregular-onset waves. It occurs in 10% of cases and is the main cause of death in patients with AMI before they enter the Coronary Unit. It can be electrical or primary and mechanical or secondary. The first is what occurs as an abrupt event in a patient with no previous evidence of circulatory insufficiency, while the mechanical is the terminal arrhythmia in a patient with severe pump failure.
2. Heart failure (pump failure) . Left ventricular failure in the course of an AMI continues to be the most important challenge for the physician in the Coronary Unit. Loss of functioning myocardium is the most important cause of heart failure in this condition, but in many cases there are other factors that contribute to triggering, maintaining or worsening ventricular failure. These include the decrease in preload due to relative or absolute hypovolemia, the increase in afterload caused by adrenergic discharge in the first hours of I AM, the presence of arrhythmias and post-infarction mechanical complications, such as ventricular dyskinesia. , or rupture of the papillary muscle or the interventricular septum.
From a clinical point of view, following the criteria of Killip and Kimball, patients are classified into four functional classes:
Class I: uncomplicated patients, without manifestations of heart failure.
Class II: minimal signs or symptoms of heart failure (basal rales, gallop, or venous hypertension).
Class III: acute lung edema due to severe
left ventricular failure.
Class IV: cardiogenic shock.
Hemodynamic monitoring in the Coronary Unit, by placing a Swan-Ganz thermodilution catheter in the capillary bed of the pulmonary artery (which represents the end-diastolic pressures of the left ventricle), is of great importance for a correct evaluation of these patients . Considering the state of pulmonary congestion and peripheral perfusion, Forrester and Ganz classify them as:
Group I: cardiac index greater than 2.2 1 / min / m2 and pulmonary capillary pressure less than 18 mm Hg. The patient is asymptomatic.
Group II: pulmonary capillary pressure is greater than 18 mm Hg. and cardiac output remains above 2.2 1 / min / m2.
The patient may be asymptomatic or present clinical and radiological signs of more or less severe pulmonary congestion. If the clinical picture is acute lung edema and the systemic resistance is very high, it may also present signs of peripheral vasoconstriction that simulate a situation of low output.
Group III: pulmonary capillary pressure is less than 18 mm Hg. and cardiac output less than 2.2 1 / min / m2. In this situation, which is absolute hypovolemia (due to vomiting, diarrhea, or excessive diuresis) or relative (in which there is no true hypovolemia, but due to decreased contractility, the ventricle needs a higher preload to maintain normal function ), the patient may be asymptomatic or present signs of peripheral hypoperfusion and arterial hypotension.
Group IV: pulmonary capillary pressure is greater than 18 mm Hg. and the cardiac index lower than 2.21 / min / m2. In this situation of severe hemodynamic deterioration, the patient practically always presents clinical signs of pulmonary congestion and low output. Blood pressure is usually low. The most serious form of heart failure, cardiogenic shock, is linked in this group. To make the diagnosis of cardiogenic shock, it is necessary that the pulmonary capillary pressure is high and thus rule out hypovolemic shock.
3. Shock cardiogenico. La Myocardial Infarc¬tion Research Unit (M.l.R.U) del National Heart and Lung Institute de Bethesda define asi al shock cardiogenico:
a) Systolic blood pressure less than 90 mm Hg, or less than 30 mm Hg. to its previous value in a hypertensive person, for at least half an hour, associated with:
b) Inadequate peripheral perfusion manifested by the following clinical biochemical criteria:
1. Urinary output less than 20 ml / hour and natriuria below 30 mEq / 1.
2. Skin cold, pale, livid, sweaty and slimy.
3. Mental disorder (cerebral hypoperfusion) characterized by agitation, drowsiness or coma.
4. Compromise of aerobic cellular metabolism (lactic acidosis).
The incidence of cardiogenic shock after AMI is approximately 15%, and mortality is overwhelming, exceeding 90% in some series. Anatomopathological studies showed that this condition occurs when more than 40% of the myocardial mass is damaged, either due to recent injury or associated with old necrosis. The extent of the damage is directly related to the prognosis and is the result of a progressive lesion, constituting a vicious cycle of ischemia, myocardial depression and increased ischemia.
Extensive necrosis of the ventricular mass causes a reduction in contractility, a decrease in cardiac output, and a decrease in blood pressure. Stroke volume falls rapidly and, if this drop reaches between 30 and 50% of its normal value, tissue hypoxemia occurs, with the consequent anaerobic cell metabolism and the development of lactic acidosis.
Through hypoxemia and the release of catecholamines (mediated by sympathetic activation), a negative feedback mechanism is installed with a consecutive increase in vascular resistance, mainly in the kidney, the splanchnic territory, and the skin, in such a way that the volume of blood returning to the right heart decreases.
This explains that, although left ventricular end-diastolic pressure increases slightly, in 75% of cases there is no pulmonary edema or increased central venous pressure.
Hypotension, norepinephrine release, and reduced effective circulating volume lead to renal and hepatic hypoperfusion. The former stimulates the renin-angiotensin-aldosterone system, and the reduction in hepatic perfusion reduces the metabolism of aldosterone in the liver. These two mechanisms produce an increase in aldosteronemia, with the consequent retention of sodium and loss of potassium at the renal level. Myocardial necrosis, anoxia, acidosis, and potassium deficiency are fertile ground for the development of serious arrhythmias.
Maintaining low cardiac output and hypoperfusion for several hours can cause greater myocardial damage, establishing a positive and irreversible feedback between decreased cardiac output and myocardial damage. The mechanism of this vicious circle seems to respond to myocardial hypoperfusion and to a humoral factor that depresses myocardial cell contractility. Hypoperfusion of the myocardium, with the consequent decrease in contractility, extends the infarcted area. Among the humoral factors, the production of active polypeptides that depress the force of myocardial contraction stands out and which are produced mainly in the pancreas and the splanchnic territory due to the rupture of lysosomes due to hypoxia of these tissues.
Thus, the vicious cycle that arises from the complications of shock, if not resolved early, leads to death quickly. Regarding the comic characteristics of this serious condition, it can be added that patients are severely compromised, with symptoms and signs secondary to poor tissue perfusion, such as: mental confusion, restlessness, vasoconstriction of integuments, cyanosis peripheral, narrow pulses, hypotension, tachycardia, oliguria (diuresis less than 20 ml / hour), etc., and pulmonary congestion: dyspnea, orthopnea, polypnea, etc. The most sensitive indicators of tissue perfusion are the state of the central nervous system and urinary flow, elements that must be evaluated permanently.
To reach the diagnosis of cardiogenic shock due to AMI, the following elements should be kept in mind:
A) prolonged chest pain in the last 48 hours;
B) sometimes acute lung edema without previous valvular heart disease, uremia or intoxication;
C) shock not due to intoxication, sepsis or hemorrhage;
D) rebel angor.
Any of the above cases must also present electrocardiographic signs of acute infarction and an increase in parasite enzymes.
A patient who retina with the above criteria, and also those mentioned in the original definition, is in cardiogenic shock.
To quantify hemodynamic disorders, their evolution and the effect of therapeutic measures, a control must be carried out with hemodynamic monitoring equipment. A very useful instrument that has already been mentioned is the Swan-Ganz catheter, which makes it possible to measure cardiac output, pressures in the pulmonary circuit and the 02 content of mixed venous blood.
4. Acute mitral regurgitation . It may be due to papillary muscle dysfunction or rupture. The incidence of rupture of a papillary muscle and / or its chordae tendineae in acute infarction is 1%. It occurs more frequently in lower infarcts, and is clinically characterized by a pansystolic murmur of mitral regurgitation radiating to the axilla, back, and left edge of the sternum. The clinical picture can range from mild left heart failure to acute pulmonary edema.
Right catheterization with Swan-Ganz shows the existence of prominent "V" waves in the monitoring of pulmonary venocapillary pressure, by retrograde transmission. The oxygen content of the right atrium and ventricle is equal, which makes the differential diagnosis with VSD. In this sense, the two-dimensional echocardiogram and, better still, the echo-Doppler, are very useful to clarify the doubtful pictures. Although the two-dimensional echocardiogram may sometimes not reveal a ruptured septal or papillary muscle, Doppler can demonstrate the resulting abnormality of flow. It should be noted that the mean survival in patients with partial rupture of a papillary muscle is three days, so that an immediate diagnosis is required with a view to surgical correction.
5. Ventricular communication (VSD) in AMI . Rupture in the interventricular septum occurs in less than 1% of patients with anteroseptal or posteroseptal AMI. Most cases occur in the first week of the acute episode. The size of the defect, and the presence or absence of a septal aneurysm determine the hemodynamic consequences. It is a complication that leads to mortality: more than 25% of cases die within the first three days, 65% in the first fifteen days, and 90% within the first two months.
Clinical findings include a palpable systolic froito and a rude holosisto1ic murmur in the left fourth or fifth intercostal space, irradiating the left sternal border, apex, and axilla. Heart failure is global. Sometimes the auscultatory difference between a murmur from VSD and that from acute mitral regurgitation can be difficult. A Swan-Ganz catheter must be placed and blood samples drawn from the superior vena cava, the right atrium, the right ventricle, and the pulmonary artery, to determine the oxygen content of the same. If the difference between the right atrium and the right ventricle is 1 or more volumes, the diagnosis of VSD is made, since a left-to-right shunt is shown. Two-dimensional echocardiography can also be used to detect and locate the post-infarction septal defect. If the VSD is small, it may not be visualized by echo, but by echo-Doppler. If the 2-dimensional echocardiogram does not find a VSD and the Doppler does not detect turbulent flow in the region of the interventricular septum, the cause of the murmur (and symptoms) will almost invariably be acute mitral regurgitation.
6. External heart rupture. This complication is responsible for around 10% of in-hospital fatal cases. It is four times more common in women, especially in patients over 60 years of age who are hypertensive. It generally occurs in the first week (third to fifth day) and is observed mainly in transmural anterior infarcts that take at least 10% of the left ventricular circumference. Clinically, it is characterized by sudden reappearance of chest pain, abrupt hemodynamic collapse, hypotension with clear signs of cardiac tamponade, and electromechanical dissociation. In this situation, while resuscitation maneuvers are being initiated, pericardiocenthesis should be attempted. If an echocardiogram is successful as soon as resuscitation begins, the presence or absence of a hemopericardium can be quickly confirmed.
7. Pericarditis . Epistenocardial pericarditis is very common (15% of cases), appearing between the first and second day, and disappearing after a week. It is characterized by persistent chest pain that may intensify with respiratory movements or changes in decubitus, radiating to the shoulders and back. The presence of a systodiastoic smear confirms the diagnosis.
The appearance of a pleural rub is common and the existence of episodes of recurrent pericarditis associated with pneumonitis may be the expression of Dressier's autoimmune syndrome, or post-infarction syndrome.
8. Post-infarction angina . Approximately 10 to 15% of patients with acute infarction will develop unstable angina within the first week of the event. Its prognosis is severe, as it is a complication that is associated with a high incidence of recurrent heart attack or death during hospitalization. In addition, the mortality of these patients is high at 3 to 6 months after the event.
This picture of recurrent angina, or early post-infarction angina, may be due to: 1) transitory increase in oxygen demand, tachyarrhythmias, anemia, drugs, changes in blood volume, or mechanical heart problems, such as ventricular aneurysm, mitral regurgitation , VSD or severe heart failure; 2) transitory drop in myocardial oxygen supply, which may respond to transient thrombosis or platelet aggregation in severely damaged arteries, or to a coronary artery spasm close to the site of an atherosclerosis stenosis.
Considering the multiple possible causes, if the patient has recurrent angina, it should be investigated why the angina recurs. A complete clinical evaluation, laboratory tests, and even a coronary angiography should be performed if the patient is expected to benefit from invasive therapy.
Even the latest angina, but which occurs within a month of the AMI and with the patient still at rest, usually responds to similar causes and its prognosis is serious, for which it is necessary to follow the guidelines mentioned above.
9. Right ventricular infarction (RV-AMI). Although isolated right ventricular AMI is very rare, this condition can be associated with a transmural infarct of the posteroinferior wall of the left ventricle.
The clinical syndrome of RV-AMI appears between 3% and 8% of posteroinferior infarcts, although it is found in between 15 and 34% of autopsies and in 30-70% of the lower infarcts studied with scintigraphy and echocardiography. These differences are due to the fact that many of the pathological lesions have no clinical or hemodynamic translation. The clinical spectrum that it originates is very wide - it can even lead to cardiogenic shock - but its diagnosis relies on clinical and hemodynamic evidence of cardiac dysfunction with a right preponderance, in the course of AMI. electrocardiographic of acute posteroinferior infarction, corroborated by enzyme elevation; 2) clinical evidence of dominant right heart failure, demonstrated by jugular engorgement, central venous hypertension greater than 18 cm H20, and 3) hemodynamic confirmation with a Swan-Ganz catheter of elevation of right ventricular end-diastole pressure or values equal to or greater than pulmonary capillary pressure. It is also accompanied by AV conduction disorders in a high percentage of cases, which in some series reaches 100%.
The presence of right failure in the course of an AMI may also respond to left ventricular failure or pulmonary thromboembolism. It corresponds to establish the differential diagnosis with these entities.
10. Pulmonary embolism . It can lead to sudden death, arrhythmias, and / or refractory heart failure in the course of an AMI. Its frequency of appearance is not known. In most cases the starting point is the deep veins of the lower limbs and the pelvis.
The clinical picture will depend on the number and size of the emboli. Thus, it can be observed from sweating, chest pain, dyspnea and moderate hypotension, to heart failure refractory to treatment, arrhythmias, shock and death. Auscultation will only reveal "a second, doubled and fixed noise, with accentuated P2. The most frequent arrhythmia is atrial fibrillation, and the ECG will show deviation of the axis to the right, clockwise rotation, right atrial enlargement and complete right bundle branch block. To confirm the diagnosis, the most useful study is the pulmonary radioisotopic scintigraphy.If this shows large areas of bilateral hypoperfusion, a pulmonary arteriography should be indicated to determine the need for an embolectomy.
11. Systemic embolisms . This is a rare complication, and is due to the formation of mural thrombi in the wall of the infarcted left ventricle or atrium, from where they migrate to the periphery. Its prognosis depends on the impacted organ (kidney, brain, lower limbs, etc.) and the size and hierarchy of the occluded artery.