
 Português
Português  Español
Español  English
English Fernando L. Soldano, Antonio Molina Rojas, Gustavo Lavenia
Síndrome caracterizada por proteinúria (sinal maiúsculo), edema, disproteinemia (hipoalbuminemia, hipergamaglobulinemia) e dislipidemia. É a consequência clínica do aumento da permeabilidade da parede capilar glomerular, portanto, é uma manifestação da doença renal glomerular. A proteinúria é considerada causadora da síndrome nefrótica se for maior que 3,5 g / 24 h / 1,73 m2 em adultos ou 40 mg / h / m2 em crianças (proteinúria na faixa nefrótica). No entanto, as manifestações clínicas da síndrome nefrótica podem não aparecer com valores superiores a este valor ou ser vistas com valores inferiores a 3,5 g / 24 h. A importância da síndrome nefrótica reside na magnitude da proteinúria e, portanto, na lesão glomerular subjacente
Fisiopatologia da proteinúria na síndrome nefrótica
A fisiopatologia central da síndrome nefrótica está no aumento da permeabilidade glomerular e na consequente perda de proteínas na urina. O resto das alterações descritas na síndrome nefrótica são uma consequência direta desta massiva proteinúria.
A parede capilar glomerular normal, composta por células endoteliais, membrana basal glomerular e células epiteliais viscerais, constitui uma barreira devido ao tamanho e à carga elétrica à passagem de proteínas maiores que 70 kd. Na síndrome nefrótica, a proteinúria poderia ser causada, pelo menos teoricamente, por um distúrbio eletroquímico (perda de eletronegatividade da barreira de filtração), ou por uma alteração estrutural da membrana de filtração que condiciona um aumento do tamanho dos poros da mesma. No tamanho dos poros da parede das células epiteliais viscerais, desempenha um papel fundamental, pois impediria a passagem de macromoléculas maiores que 150 kd. Várias moléculas de adesão, como a nefrina, bem como integrinas e proteínas do citoesqueleto, parecem ser constituintes fundamentais desses poros. Além do que, além do mais, alguns modelos teóricos sugerem a existência de outros poros com raio maior, cujo número estaria aumentado na lesão glomerular, favorecendo a proteinúria maciça. Esses poros encolheriam com drogas antiproteinúricas, como os inibidores da enzima de conversão da angiotensina (ACE). A alteração estrutural das células epiteliais glomerulares é característica de todos os processos associados à síndrome nefrótica. A capacidade proliferativa limitada dessas células após a noxa favoreceria a proteinúria e a insudação das proteínas plasmáticas, que à microscopia de luz se traduziriam em depósitos de proteínas, alterações na membrana basal glomerular e anormalidades do mesângio glomerular. Esses poros encolheriam com drogas antiproteinúricas, como os inibidores da enzima de conversão da angiotensina (ACE). A alteração estrutural das células epiteliais glomerulares é característica de todos os processos associados à síndrome nefrótica. A capacidade proliferativa limitada dessas células após a noxa favoreceria a proteinúria e a insudação das proteínas plasmáticas, que à microscopia de luz se traduziriam em depósitos de proteínas, alterações na membrana basal glomerular e anormalidades do mesângio glomerular. Esses poros encolheriam com drogas antiproteinúricas, como os inibidores da enzima de conversão da angiotensina (ACE). A alteração estrutural das células epiteliais glomerulares é característica de todos os processos associados à síndrome nefrótica. A capacidade proliferativa limitada dessas células após a noxa favoreceria a proteinúria e a insudação das proteínas plasmáticas, que à microscopia de luz se traduziriam em depósitos de proteínas, alterações na membrana basal glomerular e anormalidades do mesângio glomerular.
A barreira dependente de carga se deve às cargas negativas dos glicosaminoglicanos polianiônicos ricos em sulfato de heparano da membrana basal glomerular, que restringiriam a passagem de pequenas proteínas polianiônicas plasmáticas com tamanho entre 70-150 kd, principalmente albumina. Por esse motivo, proteínas com carga positiva, em tamanhos iguais, apresentam maior depuração renal do que proteínas com carga negativa. A nefropatia de alteração mínima é o paradigma das doenças causadas por um distúrbio glomerular eletroquímico. Nestes casos, nenhuma alteração morfológica é observada à microscopia de luz e a proteinúria é muito seletiva (a albumina e outras proteínas negativas são perdidas, enquanto as de maior peso molecular, como a IgG, são retidas).
Pequenas modificações nas propriedades de permeabilidade da parede glomerular produzem perdas significativas de proteínas de peso molecular intermediário (entre 40 e 150 kd). Estes incluem: albumina, IgG, transferrina, ceruloplasmina e glicoproteína. Pequenas quantidades de proteínas ligeiramente maiores, como pequenas formas de HDL (200 kd), também são perdidas. Proteínas de peso molecular muito alto, como IgM, macroglobulinas, fibrinogênio, fator XIII, fibronectina e lipoproteínas maiores, não são perdidas, mesmo com grandes alterações na permeabilidade e seletividade glomerular. Do ponto de vista prático, proteínas maiores que 200 kd não são perdidas na síndrome nefrótica.
Independentemente da extensão do dano glomerular, outros fatores podem condicionar uma variação muito ampla na proteinúria, como filtração glomerular, fluxo plasmático renal, atividade do sistema renina-angiotensina, produção plasmática e concentração de albumina, ingestão diária de proteína e administração de medicamentos anti-hipertensivos.
Clínica de Síndrome Nefrótica
Hipoalbuminemia
A albumina é a proteína plasmática mais abundante. A albumina filtrada é catabolizada em parte pelo túbulo renal, cuja taxa catabólica aumenta e pode representar até 20% da albumina filtrada na síndrome nefrótica.
Para compensar as perdas, o fígado aumenta a taxa de síntese de albumina em até 300% por meio de mecanismos de transcrição. Esse aumento está correlacionado com a albuminúria, mas não com a pressão oncótica plasmática ou a concentração de albumina sérica, e é abolido se a ingestão de proteínas for diminuída, o que explica por que as dietas pobres em proteínas diminuem a proteinúria, mas não aumentam a proteinúria. concentração de albumina no plasma.
A hipoalbuminemia (albumina inferior a 3 g / dl) surge quando a proteinúria e o catabolismo renal da albumina excedem a capacidade de síntese hepática. O grau de hipoalbuminemia está correlacionado com a magnitude da proteinúria, embora não de forma constante, pois outros fatores como a idade, o estado nutricional e o tipo de lesão renal também influenciam, o que justifica que haja pacientes com proteinúria muito elevada sem hipoalbuminemia. Esse achado é característico de algumas lesões glomerulares que se apresentam com hiperfiltração, como nefropatia da obesidade, nefropatia de refluxo ou secundária à redução da massa renal.
Edema
O aparecimento de edema é o sinal clínico mais visível e geralmente é o motivo da consulta, é um edema mole e depressível que se acumula em áreas de declínio (pés, sacro) e em regiões com pequena pressão tecidual, como na região periorbital. Se a hipoalbuminemia for grave, podem ocorrer ascite e derrame pleural. O edema pulmonar não ocorre a menos que haja insuficiência renal ou cardíaca.
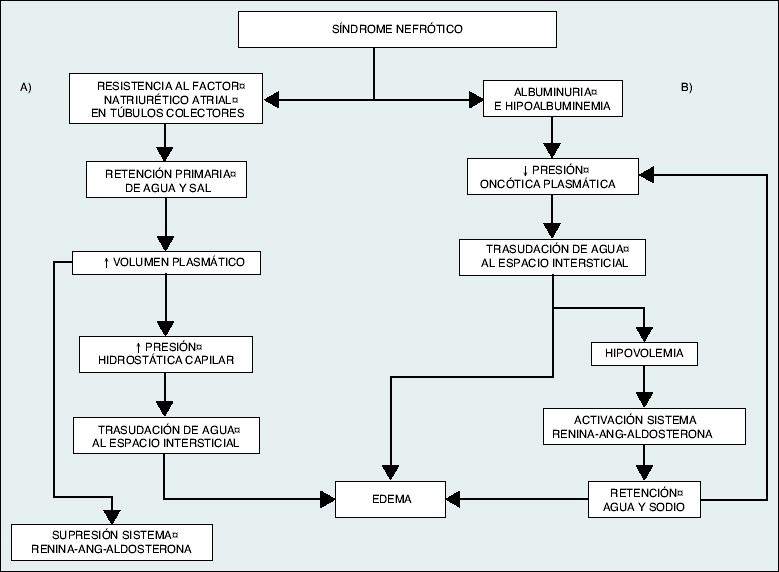
A teoria clássica é a da hipovolemia, em que a retenção renal de água e sódio é consequência da diminuição da pressão oncótica plasmática decorrente da hipoalbuminemia. Isso favoreceria o extravasamento de líquido do compartimento intravascular para o intersticial, causando edema. A resposta homeostática à hipovolemia ativaria o sistema renina-angiotensina-aldosterona, aumentaria o tônus simpático e a liberação de hormônio antidiurético. Gradualmente, o volume plasmático se normalizaria às custas de um aumento no espaço extracelular e um aumento significativo no edema.
A teoria alternativa que explicaria a origem do edema, com a teoria da expansão de volume ou hipervolemia. Diante disso, haveria dano intrínseco ao néfron que levaria a um aumento primário da reabsorção tubular de sódio, independente da situação hemodinâmica, induzindo uma expansão de volume que favoreceria o aparecimento de edema. A teoria da hipervolemia é hoje a mais aceita. No caso de hipoalbuminemia muito grave, principalmente em crianças, ocorreria a situação de hipovolemia efetiva e os mecanismos propostos pela teoria clássica ou hipovolêmica seriam mais importantes.
|
Patogênese da formação de edema na síndrome nefrótica. A) Teoria da hipervolemia ou expansão de volume, segundo a qual o edema seria o resultado da retenção de água e sódio pelo próprio rim doente, o que produziria uma expansão do volume plasmático e um aumento da pressão hidrostática capilar que, juntamente com a hipoalbuminemia , favoreceria a transudação de fluido para o espaço intersticial. B) Teoria clássica ou hipovolêmica, segundo a qual a diminuição da pressão oncótica secundária à hipoalbuminemia favoreceria um quadro hipovolêmico e a retenção de água e sódio pela ativação do sistema renina-angiotensina-aldosterona. Esta situação tem significado clínico apenas em circunstâncias de hipoalbuminemia grave (albumina <2 g / dl). |
Dislipemia
É o resultado do aumento da síntese hepática de lipídios e apolipoproteínas e, sobretudo, da diminuição do clearance de quilomícrons, lipoproteínas de muito baixa densidade (VLDL), baixa densidade (LDL) e densidade intermediária (IDL).
A hipercolesterolemia é a anormalidade lipídica mais constante e sua gravidade está inversamente correlacionada com a magnitude da hipoalbuminemia e com a diminuição da pressão oncótica plasmática. A hipertrigliceridemia, menos comum, só aparece quando a albumina sérica cai abaixo de 1–2 g / dl.
De forma muito característica, ocorre a lipidúria, com cilindros gordurosos sendo observados no sedimento urinário. É aconselhável adotar as medidas terapêuticas necessárias para normalizar a hiperlipidemia em todos os pacientes com síndrome nefrótica, como medicamentos hipolipemiantes para
Reduz a morbidade e mortalidade cardiovascular ou a progressão da doença renal com a correção de anormalidades lipídicas.
Trombose
A trombose da veia renal e, em geral, os fenômenos tromboembólicos constituem uma das complicações mais graves da síndrome nefrótica. Hipoalbuminemia (albumina sérica inferior a 2,5 g / dl), proteinúria superior a 10 g / 24 h, valores de antitrombina III inferiores a 75% do normal e hipovolemia foram associadas a um risco elevado de complicações tromboembólicas.
Infecções
Existem vários fatores que condicionam uma alta suscetibilidade a infecções em pacientes nefróticos, incluindo deficiência de IgG (devido à diminuição da síntese e aumento das perdas por vazamento e catabolismo renal), opsonização inadequada devido à diminuição do fator B do complemento, bem como distúrbios na imunidade celular favorecido pela deficiência de vitamina D, desnutrição e deficiências de transferrina e zinco, ambas essenciais para o funcionamento adequado dos linfócitos.
Hipertensão arterial
Achado comum na síndrome nefrótica. Os fatores predisponentes para hipertensão não são claramente compreendidos e podem estar relacionados à retenção de sódio e água ou à perda urinária de substâncias vasodilatadoras (hipotensivas).
Desordens endócrinas
Há um déficit relativo de EPO (eritropoietina), porém o impacto na eritropoiese é nulo, exceto em casos isolados. Alterações no metabolismo da vitamina D, diminuição das concentrações de tiroxina (T4) e triiodotironina total (T3) também podem ser encontradas, mas os pacientes são clinicamente eutireoidianos.
Outras complicações
Nas síndromes nefróticas de longo prazo, outras complicações podem ocorrer. Assim, a proteinúria persistente e o catabolismo renal aumentado de proteínas filtradas desencadeiam um balanço de nitrogênio negativo e desnutrição protéica. Anormalidades tubulares proximais, como glicosúria, hiperfosfatúria e síndrome de Fanconi, também foram descritas.
 Doenças glomerulares
Doenças glomerulares
Inicialmente, explicaremos o que é, em geral, a classificação das doenças glomerulares por tempo de evolução e depois por etiologia. Faremos referência neste capítulo, especialmente para aqueles com Síndrome Nefrótica e, em seguida, aqueles com Síndrome Nefrótica predominantemente. Uma primeira abordagem clínica permite que a glomerulonefrite seja classificada de acordo com sua evolução ao longo do tempo. A glomerulonefrite aguda evolui em dias, com um início, e muitas vezes um fim, bem definido no tempo. A glomerulonefrite rapidamente progressiva é caracterizada por uma rápida deterioração da função renal ao longo de semanas ou meses, sem tendência espontânea de melhora, com um substrato histológico comum, que é a proliferação extracapilar na forma de crescentes epiteliais.A glomerulonefrite crônica é caracterizada por seu curso insidioso e evolução variável ao longo dos anos. Outra abordagem é etiológica, visto que em muitas glomerulonefrites um fator causal pode ser identificado. Dois grandes grupos são assim distinguidos: glomerulonefrite primária e secundária. No entanto, essa classificação etiológica tem muitas limitações, pois não nos permite distinguir entre as diferentes glomerulonefrites primárias, de origem desconhecida e, além disso, a mesma causa pode produzir diversos padrões de doença glomerular de curso clínico e prognóstico diferentes. No diagrama a seguir veremos a celularidade glomerular normal e histologia, a fim de compreender as diferentes variantes de glomerulopatias que podem ser encontradas.Outra abordagem é etiológica, visto que em muitas glomerulonefrites um fator causal pode ser identificado. Dois grandes grupos são assim distinguidos: glomerulonefrite primária e secundária. No entanto, essa classificação etiológica tem muitas limitações, pois não nos permite distinguir entre as diferentes glomerulonefrites primárias, de origem desconhecida e, além disso, a mesma causa pode produzir diversos padrões de doença glomerular de curso clínico e prognóstico diferentes. No diagrama a seguir veremos a celularidade glomerular normal e histologia, a fim de compreender as diferentes variantes de glomerulopatias que podem ser encontradas. Outra abordagem é etiológica, visto que em muitas glomerulonefrites um fator causal pode ser identificado.Dois grandes grupos são assim distinguidos: glomerulonefrite primária e secundária. No entanto, essa classificação etiológica tem muitas limitações, pois não nos permite distinguir entre as diferentes glomerulonefrites primárias, de origem desconhecida e, além disso, a mesma causa pode produzir diversos padrões de doença glomerular de curso clínico e prognóstico diferentes. No diagrama a seguir veremos a celularidade glomerular normal e histologia, a fim de compreender as diferentes variantes de glomerulopatias que podem ser encontradas.
|
Classificação histológica de glomerulonefrite primária
|
|
Classificação etiológica, histológica e clínica de glomerulonefrite (GN)
GN associada a doenças sistêmicas
|
Causas da síndrome nefrótica
Qualquer doença glomerular, primária ou secundária, pode produzir síndrome nefrótica em algum momento de sua evolução. As causas mais comuns estão listadas na Tabela 1. As doenças que causam a síndrome nefrótica variam com a idade. Assim, a maioria das síndromes nefróticas em crianças é devida à nefropatia de alteração mínima. Em adultos, a causa mais comum é uma forma secundária, a nefropatia diabética. A prevalência de glomerulonefrite primária em adultos com síndrome nefrótica é variável, dependendo da região geográfica e da população estudada. As mais comuns são nefropatia membranosa, glomerulonefrite esclerosante e focal (crescente e a principal causa em negros) e nefropatia de alteração mínima.
| Etiologia da síndrome nefrótica | ||
| Glomerulonefrite primária | Crianças (%) | Adultos (%) |
| Nefropatia de mudança mínima | 52,2 | 14,8 |
| Glomerulonefrite esclerosante e focal | 33,3 | 15,1 |
| Glomerulonefrite Membranosa | 5,8 | 22,2 |
| Glomerulonefrite mesangiocapilar | 4,3 | 7 |
| Nefropatia IgA | - | 4,9 |
| Outras lesões glomerulares primárias | - | 10,3 |
|
Doenças glomerulares secundárias
|
|
Avaliação clínica e laboratorial da síndrome nefrótica
|
Nefropatias glomerulares
Nefrose lipóide ou alterações mínimas:
É uma doença típica da infância (80%), embora possa ocorrer em qualquer idade. Clinicamente, caracteriza-se por alterações da síndrome nefrótica pura, com proteinúria seletiva. Embora geralmente seja idiopática, pode, excepcionalmente, estar associada a outras doenças. Pouca alteração funcional, exceto pela sua apresentação em idades avançadas, onde pode aparecer insuficiência renal oligúrica, determinada por hipovolemia. Histologicamente, é caracterizada pela ausência de lesões histológicas relevantes à microscopia de luz e pela presença de fusão de podócitos à microscopia eletrônica. Na imunofluorescência, embora geralmente negativa, pode-se observar algum depósito mesangial de IgM ou complemento, acompanhado ou não de proliferação mesangial mínima.
Glomeruloesclerose segmentar e focal:
Pode ocorrer em adultos e crianças, pode ser primário e secundário. É caracterizada pela presença de lesões esclerosantes em alguns glomérulos (focais) e apenas em algumas alças capilares (segmentares) e depósitos imunes de IgG e C3. Clinicamente, apresentam síndrome nefrótica com proteinúria não seletiva, microhematúria, leucocitúria, hipertensão arterial e graus variáveis de comprometimento da função renal.
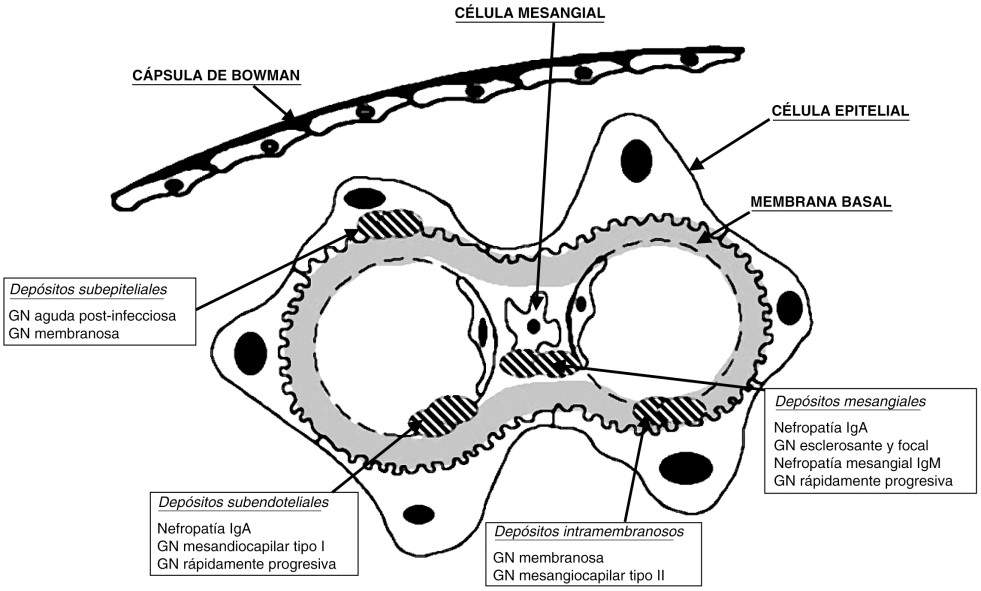
Glomerulopatía Membranosa:
Clinicamente, apresenta proteinúria intensa e é a causa mais comum de síndrome nefrótica em adultos. Pode ser primária e secundária, nas formas secundárias, pode estar associada aos vírus das Hepatites B, C e neoplasias. Do ponto de vista morfológico, caracteriza-se pela existência de depósitos subepiteliais ao longo da membrana basal na ausência de proliferação celular, o que confere a característica histológica de espessamento difuso da membrana basal. O referido depósito é gradativo e dá a classificação morfológica em 4 etapas. Entre 20-30% dessa glomerulopatia é autolimitada. À medida que a lesão progride, os glomérulos tornam-se esclerosados e ocorrem fibrose intersticial e atrofia tubular, levando a IRC em anos.
Glomerulopatía Membranoproliferativa:
Também conhecida como mesangiocapilar, é uma doença glomerular crônica, podendo ser primária ou secundária, com curso clínico variável. Nas formas secundárias, encontram-se como fatores as doenças infecciosas, hereditárias ou multissistêmicas (LES, Hepatites, neoplasias, crioglobulinemia, etc.). A característica predominante é a proliferação mesangial, juntamente com espessamento da parede capilar glomerular e hipocomplementemia (característica de diminuição de C3). Existem 3 tipos de acordo com a classificação histológica: Tipo I, forma clássica, com depósitos subendoteliais; tipo II, com depósitos intramembranosos densos; Tipo III, depósitos subendoteliais e subepiteliais.A apresentação clínica não difere nos subtipos, predomina a síndrome nefrótica (50%) associada a hematúria e / ou alterações urinárias assintomáticas.
Glomerulopatia proliferativa mesangial difusa:
Incomum, histologicamente caracterizada por lesões mesangiais inespecíficas (aumento difuso da celularidade mesangial e lesões de esclerose glomerular) e clinicamente se apresenta com síndrome nefrótica com microhematúria. Na imunofluorescência, podem ser encontrados depósitos de IgM, IgA e C3.
Glomerulonefrite de evolução rápida:
É definida clinicamente pela rápida deterioração da função renal em semanas ou meses, com pouca tendência à recuperação espontânea. A característica histológica é a proliferação extracapilar em forma de crescentes de mais de 50%, sua incidência é variável e pode ocorrer em qualquer idade. Existem formas primárias (idiopáticas, com 3 tipos) e secundárias a doenças autoimunes e infecciosas.