
 Português
Português  Español
Español  English
English Fernando L. Soldano, Antonio Molina Rojas, Gustavo Lavenia
Síndrome caracterizada por hematúria macro ou microscópica (sinal maiúsculo), hipertensão arterial (devido à retenção de H 2 O e sódio), proteinúria na faixa não nefrótica (com ou sem edema), e achados no sedimento urinário, de cilindros hemáticos . Além disso, pode se manifestar clinicamente com insuficiência renal e, ocasionalmente, oligúria. A síndrome nefrítica aguda ocorre devido a várias entidades morfológicas que manifestam inflamação glomerular difusa. Sua apresentação pode ser aguda ou crônica, na forma aguda geralmente ocorre após infecção de vias aéreas superiores ou pele. Nas formas crônicas, tem início insidioso, com comprometimento progressivo da função renal, acompanhado de proteinúria, hematúria e hipertensão arterial.
Hematúria : indica dano glomerular, pode ser assintomático, micro ou macroscópico. O aparecimento de moldes hemáticos sugere inflamação glomerular de qualquer etiologia. O achado de até 1 cilindro hemático é diagnóstico da síndrome.
Hipertensão Arterial : a alteração da filtragem, retém sódio e água, provoca expansão do volume plasmático, elevando a pressão arterial, como primeiro mecanismo e também é causada pela ativação do sistema renina-angiotensina-aldosterona. O aparecimento da encefalopatia hipertensiva geralmente ocorre nas formas agudas.
Proteinúria : presente em quase todas as glomerulopatias, embora em graus variáveis. Se for leve, é assintomático, em casos graves (> 3,5 g / 24 h). Será clinicamente evidenciado com edema.
Edema : generalizado ou localizado (por exemplo, periorbital e sacro em decúbito).
Insuficiência renal : indicativo de uma queda severa na taxa de filtração glomerular, aumento da uremia e creatininemia.
Oligúria : ocorre em algumas formas agudas e é característico da de evolução rápida, levando a uma síndrome urêmica.
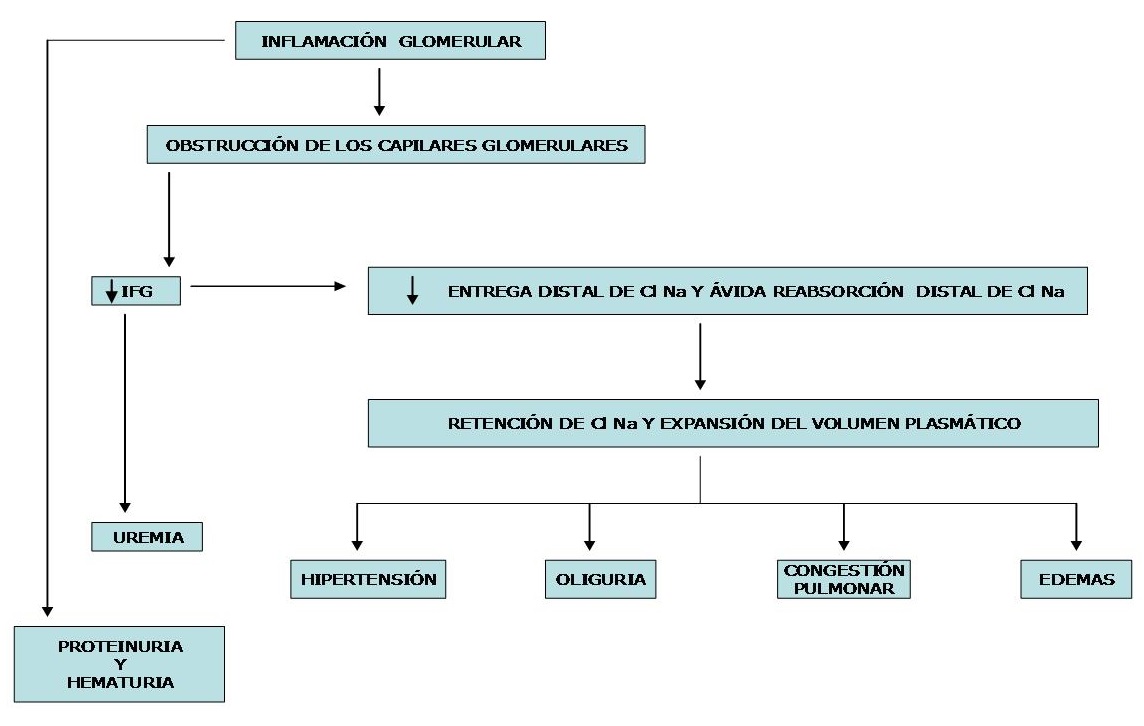
O diagrama a seguir mostra a evolução que leva a manifestações clínicas em um paciente com hematúria e diminuição de início abrupto da TFG.
Fisiopatologia:
A maioria das glomerulonefrites são mediadas imunologicamente por:
a) Imunocomplexos (IC): um antígeno, que pode ser exógeno ou endógeno, liga-se ao seu anticorpo no glomérulo ou ambos são ali depositados, após acoplamento na circulação.
b) anticorpos anti-membrana basal glomerular (anti-MBG): o anticorpo que é uma IgG endógena, liga-se à membrana basal que é o antígeno.
Em ambos os tipos de glomerulonefrite, a reação antígeno-anticorpo ativa os componentes do complemento causando quimiotaxia, citólise e aumento da permeabilidade vascular. Os neutrófilos liberam enzimas proteolíticas e aminas vasoativas das plaquetas. As células mesangiais proliferam e os linfócitos causam citólise. A membrana basal tende a engrossar, os podócitos se fundem, se formam trombos e o material celular acidofílico é depositado, obliterando o glomérulo. Essa hialinização (esclerose) é irreversível. Prostaglandinas, leucotrienos, serotonina, histamina, angiotensina II, etc. participam da flogose.
As respostas são variáveis: se poucos glomérulos são afetados, GN é focal; se todos ou a maioria são alterados, é generalizado. Se os glomérulos afetados forem apenas parcialmente, a GN é segmentar; é difuso quando eles estão em sua totalidade. A imunofluorescência detecta imunoglobulinas, componentes do complemento, fibrina, properdina, etc. Quando estes conseguem entrar no espaço de Bowman, estimulam a migração e proliferação de monócitos e células epiteliais, formando crescentes que causam o quadro de GN rapidamente evolutivo.
Vários modelos experimentais para IC (Immunocomplexes) foram descritos que confirmam a patogênese da GN humana. Uma é a nefrite por doença do soro: um coelho é injetado com albumina bovina e entre 8 a 14 dias apresenta lesões articulares, cardíacas e renais. O animal desenvolveu anticorpos que formam IC com a albumina bovina, estes se depositam no glomérulo produzindo intensa proliferação celular. O coelho sofre de hipertensão, proteinúria, hematúria e oligúria que tendem a se resolver espontaneamente. As respostas histológicas e clínicas são variáveis, com lesões agudas, crônicas, proliferativas, exsudativas, membranosas e esclerosantes.A outra nefrite experimental é a nefrite de Massugi, na qual um coelho é injetado com um extrato de rim de rato, resultando na formação de anticorpos anti-rins de rato. Se o soro desse coelho for injetado em outro rato, ele passa por uma GN (fase heteróloga). Steblay demonstrou que o coelho desenvolve uma IgG anti-glomerular e da membrana basal pulmonar. Numa segunda fase, o rato reage formando anticorpos contra Ig de coelho. Isso causa um segundo episódio glomerulonefrítico (fase autóloga). Em humanos, a síndrome de Good - Pasture de etiologia desconhecida é semelhante ao modelo de Steblay devido à sua GN e lesões pulmonares.Em alguns casos, GN Focal e em outros há hemorragias pulmonares graves e crescentes que obstruem o espaço de Bowman, causando oligoanúria. o rato reage formando anticorpos contra Ig de coelho. Isso causa um segundo episódio glomerulonefrítico (fase autóloga). Em humanos, a síndrome de Good - Pasture de etiologia desconhecida é semelhante ao modelo de Steblay devido à sua GN e lesões pulmonares. Em alguns casos, GN Focal e em outros há hemorragias pulmonares graves e crescentes que obstruem o espaço de Bowman, causando oligoanúria. o rato reage formando anticorpos contra Ig de coelho. Isso causa um segundo episódio glomerulonefrítico (fase autóloga).Em humanos, a síndrome de Good - Pasture de etiologia desconhecida é semelhante ao modelo de Steblay devido à sua GN e lesões pulmonares. Em alguns casos, GN Focal e em outros há hemorragias pulmonares graves e crescentes que obstruem o espaço de Bowman, causando oligoanúria.
|
Causas da Síndrome Nefrítica
|
Glomerulonefrite primária:
a- IgA mesangial nephropathy (IgA):
Nomeado pela descoberta de IgA como um depósito. IgAN é a glomerulonefrite primária mais comum na maior parte do mundo, mas com algumas diferenças geográficas. As campanhas de detecção de doenças renais e a política de biópsia renal explicam em grande parte essas diferenças epidemiológicas. Por razões desconhecidas, é muito raro na raça negra. Em adultos, é a principal causa de doença renal biopsiada. As nefropatias por IgA podem ser primárias e secundárias. Nas primárias, a forma idiopática (doença de Berger) é encontrada sem manifestações sistêmicas e com manifestações sistêmicas (púrpura de Scholein-Henoch).Dentre as formas secundárias, pode estar relacionada a doenças autoimunes (LES, doença de Reiter, Psoríase, AR, Wegener, Crioglobulinemia Mista, etc.), doenças infecciosas (HIV, TBC,
Clínica
É mais comum em homens, que constituem cerca de 74% de todos os casos. 80% dos casos são diagnosticados entre os 15 e os 65 anos (adultos). Na anatomia patológica, o OM apresenta aumento da matriz mesangial e hipercelularidade mesangial.
As formas clínicas mais comuns são as seguintes: distúrbios urinários assintomáticos: Ou seja, microhematúria associada à proteinúria não superior a 1 gr / 24 horas. Também hematúria macroscópica, síndrome nefrótica e raramente insuficiência renal aguda.
Diagnóstico
O diagnóstico deve ser suspeitado diante dos quadros mais típicos (anormalidades urinárias ou hematúria isolada ou recorrente), com estudo urológico normal (radiografias, ultrassonografia renal e cistoscopia), na ausência de infecção urinária.
b- Glomerulonefrite proliferativa mesangial:
É caracterizada por apresentar aumento da matriz mesangial e hipercelularidade mesangial com parede capilar normal e depósitos densos mesangiais variáveis à microscopia de luz. São classificados quanto à imunofluorescência de acordo com o predomínio de depósitos, em GN por IgM, GN por IgG, GN por C3, GN por C1q, ou então não serão encontrados depósitos e apenas proliferação. Podem assumir formas focais ou difusas e existem variedades primárias e secundárias (LES, AR). As formas clínicas mais comuns são as seguintes: distúrbios urinários assintomáticos: isto é, microhematúria associada a proteinúria de não mais de 1 gr / 24 horas. Também hematúria macroscópica, síndrome nefrótica e raramente insuficiência renal aguda.
c- Glomerulonefrite mesangiocapilar (Membranoproliferativa):
Dentre todas as glomerulonefrites, a mesangiocapilar é uma das menos frequentes. A glomerulonefrite mesangiocapilar classicamente mesangiocapilar (GNMC) foi classificada como idiopática e secundária, sendo a primeira subdividida nos tipos 1, 2 e 3 de acordo com a histologia. Recebe diferentes nomes, também chamados de membranoproliferativa, hipocomplementêmica ou lobular.
Anatomia patológica:
A característica comum dos diferentes tipos de CMNG é um aumento da matriz mesangial, proliferação de células mesangiais e espessamento da parede capilar.
Aqui, descrevemos os achados histológicos mais típicos dos três tipos de glomerulonefrite:
GNMC Tipo 1:
Glomerulonefrite mesangiocapilar com depósitos subendoteliais ou clássicos
Microscopia óptica:
A característica histológica mais comum é a hipercelularidade endocapilar e espessamento difuso da parede capilar. Essa hipercelularidade se deve tanto à presença de células mesangiais quanto de leucócitos, principalmente neutrófilos e, em menor proporção, eosinófilos. O espessamento da parede se deve à interposição da matriz mesangial e polimorfonuclear, o que dá à parede uma aparência de duplo contorno. Além disso, ocorre diminuição da luz capilar com a presença de "trombos hialinos", que não são trombos verdadeiros, mas agregados de imunocomplexos.
GNMC focal
É uma variante do tipo 1, onde as alterações citadas aparecem apenas em alguns glomérulos e seu prognóstico é relativamente favorável.
GNMC Tipo 2
Glomerulonefrite mesangiocapilar com depósitos na membrana basal ou doença de depósito denso
Microscopia óptica:
Assim como no tipo 1, proliferação mesangial, observa-se aumento da matriz mesangial e espessamento da membrana basal, mas em grau mais variável, podendo simular outros tipos de glomerulonefrite, sendo fundamental a microscopia eletrônica e a imunofluorescência. no seu diagnóstico. A lesão típica é uma alteração estrutural da membrana basal às custas de um material densoeletrônico de composição desconhecida e nem sempre visível na óptica, portanto, para diagnosticar essa nefrite, é necessária a observação em microscopia eletrônica.
GNMC Tipo 3:
Glomerulonefrite mesangiocapilar mista (membranosa e proliferativa)
É uma variante do tipo 1, em que se observam alterações na parede capilar semelhantes às do membranoso. A imunofluorescência mostra depósitos granulares de C3, IgG e IgM, principalmente na parede capilar.
Clínica
GNMC pode apresentar qualquer síndrome clínica de doença glomerular, sendo as mais frequentes anomalias urinárias assintomáticas (proteinúria não nefrótica e microhematúria) e síndrome nefrótica, que representam 30% e 50%, respectivamente. A GN mesangiocapilar atinge todas as faixas etárias, embora seja mais frequente entre 5 e 30 anos, sendo excepcional a partir da sétima década de vida. De acordo com o tipo histológico, há características clínicas distintas: o tipo 1 está associado a infecções com hipocomplementemia e idade média de início em torno de 24 anos. O tipo 2 afeta pacientes mais jovens com maior incidência de insuficiência renal aguda e nefrite rapidamente progressiva. O tipo 3 é freqüentemente assintomático, sendo mais freqüente o achado de microhematúria e proteinúria.
d- Glomerulonefrite extracapilar:
A glomerulonefrite com proliferação extracapilar é caracterizada pela existência de um acúmulo de células em forma de "crescentes" que deslocam e ocupam as estruturas normais da espiga glomerular. Essa proliferação é composta por células com aspecto epitelial, localizadas na região central da espiga glomerular, motivo que justifica o nome de proliferação extracapilar. Essas crescentes são compostas por vários tipos de células, principalmente macrófagos, que passaram para o espaço de Bowman através da parede capilar glomerular rompida e células epiteliais na camada parietal da cápsula de Bowman. A lesão inicial que desencadeia a formação de crescentes está na ruptura da membrana basal glomerular.Essa ruptura permite a passagem de fibrina e monócitos para o espaço de Bowman. Em síntese, a sequência patogênica seria a seguinte: 1) depósito de anticorpos, imunocomplexos e complemento que danificam a parede capilar, em um processo no qual as células mononucleares e a imunidade celular parecem ter papel fundamental, 2) passagem da fibronectina e de fibrina para o espaço urinário, 3) atração de monócitos circulantes, 4) proliferação de células epiteliais da cápsula de Bowman, 5) síntese aumentada de proteínas da matriz com formação de crescentes e 6) evolução para fibrose, devido ao aumento da síntese de colágeno e fibroblastos infiltração.
Glomerulonefrite rapidamente progressiva define, em termos clínicos, um grupo de doenças glomerulares caracterizadas por: 1) a presença de crescentes em mais de 50% dos glomérulos, excluindo os completamente esclerosados, e 2) um curso clínico caracterizado por deterioração progressiva e rápida função renal, de modo que, na ausência de tratamento, cerca de 85% dos pacientes atingem insuficiência renal em estágio final em poucos dias, semanas ou meses. Quando o percentual de crescentes é inferior a 50%, a evolução é mais favorável e, caso contrário, não atribuível à proliferação extracapilar. Outras entidades que apresentam quadro clínico semelhante são: algumas formas de necrose tubular aguda, nefrite intersticial imunoalérgica aguda, síndrome hemolítico-urêmica, doença ateroembólica e mieloma renal.
|
Classificação de glomerulonefrite extracapilar
|
Clínica
Ela aparece em pacientes de meia-idade ou adultos, e igualmente em ambos os sexos. A síndrome de Goodpasture é mais comum em homens jovens. Às vezes, há artralgias, mialgias ou uma síndrome de gripe. Alguns fatores ambientais são os gatilhos de todo o quadro clínico, como tabaco, cocaína, infecção respiratória, edema pulmonar e exposição a hidrocarbonetos. O envolvimento renal é caracterizado por oligúria, hematúria ou ambas. A pressão arterial está normal ou ligeiramente elevada. Ocasionalmente, ocorre hemorragia alveolar assintomática que é diagnosticada após o achado de macrófagos carregados de hemossiderina no escarro ou por aumento da captação pulmonar de CO.Como achado laboratorial, a proteinúria geralmente não é nefrótica e a hematúria de origem glomerular é praticamente constante. A filtração glomerular diminui progressivamente muito rapidamente. O complemento sérico é normal. A anemia ferropriva ocorre em casos de hemorragia alveolar. 95% dos pacientes apresentam níveis elevados de anticorpos IgG α-MBG, detectados com RIA ou ELISA. Alguns antígenos do sistema de histocompatibilidade principal são mais comuns do que na população saudável (HLA-DR2, HLA-DR4 e outros).
Glomerulonefrite secundária pós-infecciosa:
Glomerulonefrite endocapilar aguda.
A glomerulonefrite endocapilar é caracterizada pelo crescimento celular difuso na espiga glomerular. A hipercelularidade glomerular é devida à proliferação, especialmente de células mesangiais, e à infiltração de células inflamatórias. A apresentação clínica é variável. A apresentação clássica é a síndrome nefrítica aguda, mas às vezes pode se manifestar apenas com hematúria microscópica e vários graus de proteinúria; mais raramente, pode se manifestar com síndrome nefrótica. Fisiopatologicamente, existe um estímulo antigênico endógeno ou exógeno e uma resposta imune que toma o glomérulo renal como órgão de choque. Dependendo da etiologia, o sistema complemento é ativado. Além disso, há uma ativação local da cascata de coagulação.
A glomerulonefrite aguda pós-estreptocócica é o exemplo típico de glomerulonefrite endocapilar aguda.
Glomerulonefrite aguda pós-estreptocócica (GNAPE)
O quadro clínico se manifesta 10 a 15 dias após uma dor de garganta vermelha, devido ao estreptococo serótipo M 1, 3, 4, 6, 12, 25 e 49 ou após um Pioderma devido ao serótipo M 2, 31, 49, 51, 52 , 55, 56, 57, 59, 60 e 61. A lesão é proliferativa e difusa, com neutrófilos abundantes.
As manifestações clínicas são hematúria, edema, hipertensão, oligúria, fraqueza, anorexia e, ocasionalmente, dor incômoda na fossa lombar. O quadro clínico completo da síndrome nefrítica aguda (hematúria, edema e hipertensão arterial) ocorre em 50% dos casos de GNAPE sintomática, mas algumas dessas manifestações ocorrem em 95% dos casos clínicos. Assim, os termos glomerulonefrite aguda e síndrome nefrítica são às vezes usados indistintamente. Menos de 4% dos pacientes têm proteinúria na faixa nefrótica (> 3,5 g / dia) e <1% tem uma elevação rapidamente progressiva dos produtos de nitrogênio.
Geralmente melhora espontaneamente, os pacientes nos quais a proteinúria persiste ou recorre, evoluem para insuficiência renal crônica.
Glomerulonefrite multissistêmica secundária:
Glomerulonefrite hereditária de Alport:
É uma doença autossômica dominante associada à surdez e distúrbios oculares. A glomerulonefrite é focal e proliferativa com inflamação intersticial, fibrose e células espumosas. O maior percentual de aparecimento está entre 5 e 20 anos de idade e clinicamente se apresenta com hematúria, proteinúria, hipertensão e ocasionalmente síndrome nefrótica.
Glomerulonefrite lúpica:
Apresenta-se clinicamente com quadro de fadiga, fraqueza generalizada, anorexia, emagrecimento, febre, artralgia, eritema malar, dermatose e alopecia. O glomérulo pode mostrar depósitos de HF com histologia normal, focal ou proliferativa generalizada, membranosa ou de evolução rápida.
Vasculite Sistêmica:
Eles constituem um grupo de entidades clínicas que afetam (inflamação e necrose predominantemente um tipo e tamanho de vasos e órgãos. Entre eles estão:
- Granulomatose de Wegener: causa GN focal ou de evolução rápida, apresenta-se com envolvimento do trato aéreo superior e inferior, como epistaxe, rinorréia, sinusite e deformação do septo nasal, cavitação pulmonar, tosse, dispneia e manifestações gerais (síndrome geral): febre alta, declínio geral, anorexia e perda de peso.
- Panarterite nodosa (PAN): São acometidas artérias médias e pequenas, essencialmente rim, coração e sistema nervoso central, apresenta-se com uma síndrome geral e geralmente respeita o pulmão, Envolvimento cardíaco (70%) na forma de angina, infarto ou pericardite . Envolvimento renal (80%) com proteinúria, hematúria, cilindros celulares e IR progressiva.
- PAN microscópica: afeta pequenas artérias, está associada com G. de Wegener e glomerulonefrite necrotizante idiomática. Clinicamente, apresenta-se com GN de evolução rápida.
- Glomerulonefrite de Schonlein-Henoch: é focal ou de evolução rápida. Está associada a púrpura, artrite e cólicas abdominais.
- Doença de Churg-Strauss: granulomatose alérgica e vasculite, apresenta manifestações alérgicas, rinite, asma e tipicamente eosinofilia no sangue periférico. 70% apresentam manifestações cutâneas (nódulos subcutâneos nas extremidades, púrpura palpável, rash maculopapular e petéquias). Envolvimento renal com GN focal e segmentar.
Crioglobulinemia mista essencial:
GN pode ser proliferativa ou membranosa e pode estar associada a púrpura, artralgias e fenômenos de Raynaud.
Anemias hemolíticas microangiopáticas: SHU (síndrome hemolítico-urêmica) e TTP (púrpura trombocitopênica trombótica)
Eles são entidades diferentes, que compartilham mecanismos patogênicos comuns e manifestações clínicas. Eles se apresentam com anemia hemolítica microangiopática, trombocitopenia e IRA. A SHU é mais frequente em crianças, precedida de episódio de diarreia, entre 40% e 80% têm IRA e 90% têm hematúria e proteinúria. A PTT, mais frequente em adultos, na terceira e quarta décadas de vida, costuma ser acompanhada de febre e manifestações neurológicas.