
 Português
Português  Español
Español  English
English Julio Libman, Astrid L. Libman
A síndrome de hiperplasia adrenal congênita, cuja expressão clínica varia de acordo com suas diversas formas, deve-se à deficiência total ou parcial de algumas das enzimas envolvidas na síntese de esteróides adrenais, com diminuição na produção de esteróides distais ao bloqueio e aumento de alguns anteriores a ela, por acúmulo e desvio da síntese para vias metabólicas alternativas.
Fisiopatologia
A hiperplasia adrenal congênita constitui um grupo de alterações autossômicas recessivas na esteroidogênese adrenal.
A síntese de esteróides adrenais ocorre a partir do colesterol pela ação de várias enzimas (Fig. 66-1). Sua produção e secreção, especialmente de glicocorticóides e esteróides de ação sexual, e em muito menor grau de aldosterona, são reguladas por centros do SNC, que controlam a produção do hormônio liberador de ACTH hipotalâmico, um peptídeo de 41 aminoácidos que, sistema portal hipotálamo-hipófise, atinge a adenohipófise, onde determina a liberação de ACTH, um peptídeo de 39 aminoácidos cujo principal efeito fisiológico é estimular a síntese de cortisol. A secreção de ACTH, que ocorre episodicamente, e a secreção de cortisol estão sujeitas ao que se conhece como ritmo circadiano, com valores máximos nas primeiras horas da manhã e mínimos nas últimas horas do dia.
O bloqueio da síntese de cortisol, devido à falta total ou parcial de qualquer uma das enzimas envolvidas em sua síntese, provoca diminuição de seus níveis plasmáticos, aumento da produção de ACTH e hiperplasia adrenal bilateral. Quando a deficiência enzimática é marcada, a produção de cortisol não é normalizada, mas a dos precursores com ação androgênica ou de retenção de Na + aumenta significativamente, o que determina uma série de manifestações clínicas variáveis de acordo com a localização do bloqueio enzimático.
Sintomas e sinais
As manifestações do quadro clínico variam de acordo com a deficiência enzimática presente.
Deficiência de 21-hidroxilase . É o tipo mais comum de hiperplasia adrenal congênita. Existem duas variedades: a) a forma simples eb) a forma perdedora de sal.
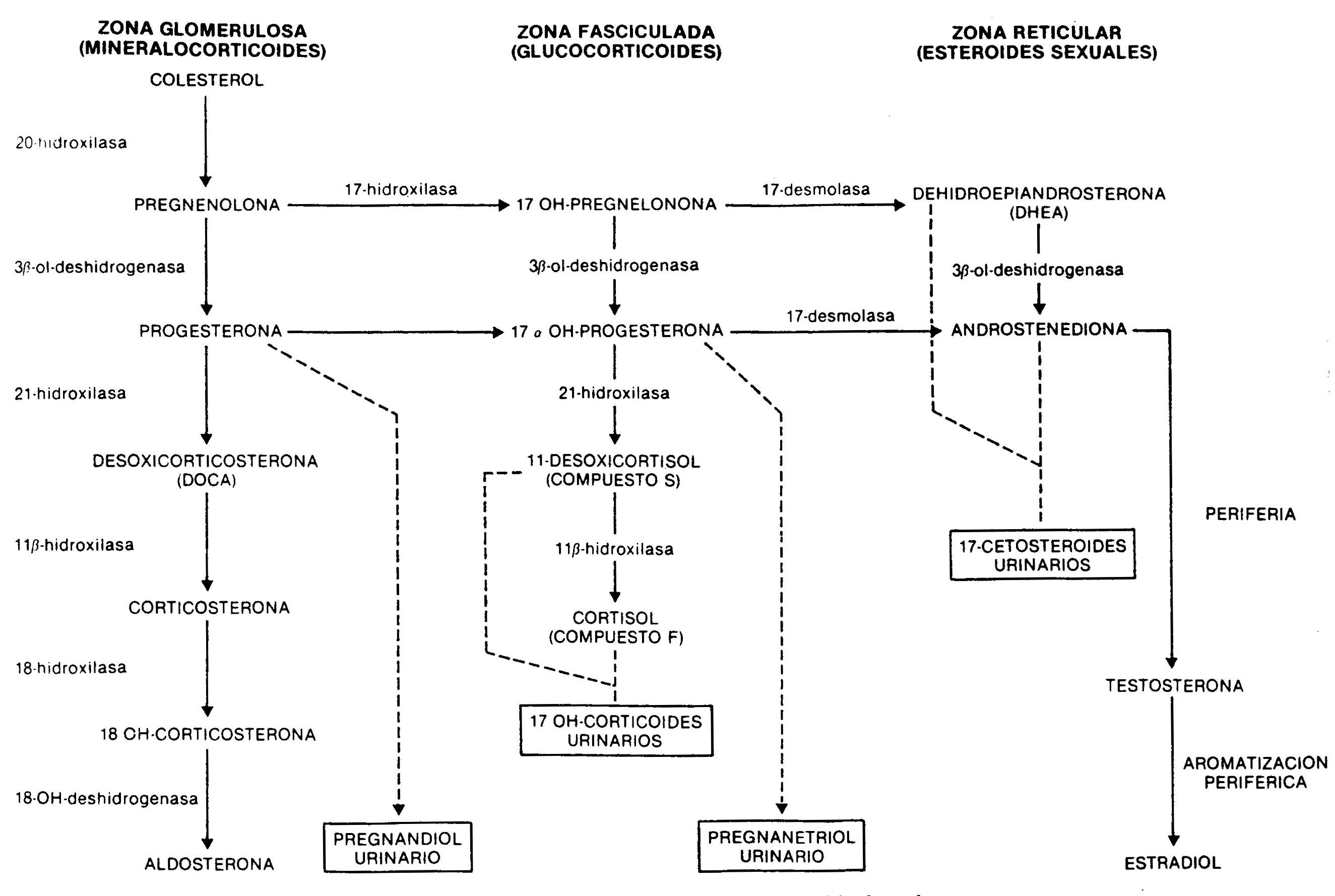
Fig. 66.1 - Síntese de esteróides adrenais a partir do colesterol
a) Na variedade simples há uma deficiência parcial bem compensada pelo aumento do ACTH, de forma que a produção de cortisol se aproxima da normalidade. O aumento do ACTH produz uma elevação acentuada dos andrógenos e precursores do cortisol. Androstenediona, altamente aumentada no sangue, é um andrógeno fraco que é metabolizado perifericamente em testosterona; isso causa masculinização da genitália externa durante a vida fetal no paciente genotipicamente feminino, com aumento do clitóris e vários graus de fusão dos grandes lábios, que podem mimetizar um homem com criptorquidia e hipospádia. Nos homens, não há anormalidades na genitália externa ao nascimento.Posteriormente, ocorre uma virilização progressiva em ambos os sexos, com aparecimento de pelos axilares e púbicos, faciais, alterações na voz e aumento da massa muscular. Mesmo quando na primeira infância são mais altos do que o correspondente à idade e ao sexo, a fusão precoce das epífises devido à ação androgênica leva à baixa estatura final. Na puberdade, e na ausência de tratamento adequado, o aumento fisiológico das gonadotrofinas não ocorre devido a uma inibição pelos esteróides sexuais circulantes, e o resultado disso é amenorréia primária nas mulheres e testículos pequenos nos homens. Na forma simples e não perdedora do sal, não há dificuldades no equilíbrio da hidrossalina e a pressão arterial é normal.17-cetosteroides, 17OH-progesterona e, em menor grau, pregnandiol estão aumentados, diminuindo seus valores na urina após a administração por 7 dias de 1,
b) Na forma perdedora de sal, há uma deficiência completa de 21-hidroxilase. Mesmo quando há aumento compensatório do ACTH, os níveis de cortisol são baixos, assim como a aldosterona, de forma que os sinais de insuficiência adrenal aguda aparecem na primeira semana de vida, com anorexia, vômitos, perda de peso, diarreia, desidratação e hipercalemia. Se não for tratado, o colapso vascular se desenvolve e a morte segue.
O diagnóstico pré-natal da deficiência de 21-hidroxilase pode ser feito no primeiro trimestre da gravidez por análise genética molecular das células das vilosidades coriônicas e quantificação de 17-hidroxiprogesterona em uma pequena quantidade de líquido amniótico.
Deficiência de 11-beta-hidroxilase . Causa uma deficiência na síntese de cortisol com hipersecreção de ACTH. A altura do bloqueio determina a produção excessiva de precursores do cortisol, como o 11-desoxicortisol e seu metabólito tetrahidro-derivado, que são quantificados na urina como corticosteroides 17OH. Conseqüentemente, eles estão elevados, assim como a 17OH-progesterona e seu metabólito, pregnanetriol. A deficiência também limita a conversão da desoxicorticosterona em corticosterona, com o conseqüente acúmulo desse potente mineralocorticoide, responsável pela hipertensão arterial observada nessa forma de hiperplasia adrenal congênita. Como consequência do déficit enzimático, ocorre também o curto-circuito para a síntese de andrógenos.A deficiência de 11-hidroxilase é, portanto, caracterizada por
Deficiência de 20-hidroxilase ou colesterol desmolase . É uma forma rara que afeta a conversão do colesterol em pregnenolona; consequentemente, há alteração na produção de glicocorticóides, mineralocorticóides e esteróides sexuais. A falta da primeira determina quadro grave de insuficiência adrenal, com perda de Na +, desidratação, hipovolemia e hipotensão. A deficiência de andrógenos é a causa de pacientes afetados com genitália externa feminina, seja seu cariótipo XX ou XY.
Deficiência de 3-beta-ol-desidrogenase . Há um defeito na conversão de pregnenolona em progesterona, de 17OH-pregnenolona em 17OH-progesterona e de desidroepiandrosterona em androstenediona. Como consequência, ocorre diminuição dos mineralocorticóides, glicocorticóides e testosterona, o que causa insuficiência adrenal com genitália ambígua nos homens e hipertrofia clitoriana moderada nas mulheres, devido ao acúmulo de dehidroepiandrosterona, um androgênio fraco. Este defeito também está presente na gônada.
Deficiência de 17-hidroxilase . Essa deficiência enzimática resulta na incapacidade total ou parcial de sintetizar cortisol e esteróides sexuais. Como visto na Figura 66-1, essa enzima não está envolvida na produção de mineralocorticóides. A produção de desoxicorticosterona, controlada pelo ACTH, está muito aumentada, resultando em hipertensão arterial leve, retenção de Na +, expansão do volume plasmático e inibição do sistema renina-angiotensina. O aumento da produção de corticosterona compensa a menor síntese de cortisol. Os machos apresentam genitália ambígua devido à diminuição da produção de testosterona. A deficiência enzimática também está presente nos ovários, com a conseqüente falta de desenvolvimento sexual na puberdade.
Metodologia de estudo
Do exposto, pode-se inferir que pode-se suspeitar da existência de hiperplasia adrenal congênita em recém-nascido com genitália externa ambígua, associada ou não a vômitos, desidratação e colapso circulatório ou ao desenvolvimento de hipertensão. Em um homem idoso, pode apresentar crescimento acelerado e pseudopuberdade precoce, ou seja, desenvolvimento de características sexuais secundárias com testículos infantis.
Na presença de algumas dessas manifestações, a cromatina nuclear deve ser avaliada em um esfregaço de células da mucosa bucal e em leucócitos circulantes para determinar o sexo cromossômico e quantificar 17-cetosteroides urinários, corticosteroides 17OH e pregnanetriol, e 17OH- progesterona e ionograma sanguíneo, juntamente com 11-desoxicortisol, em caso de hipertensão concomitante. A idade óssea é avançada em imagens radiográficas de mão e punho.